





Abstract
Acute respiratory distress syndrome (ARDS) is associated with altered plasma and lung iron chemistry. Iron can promote microbial virulence and catalyse pro-oxidant reactions, thereby contributing to the oxidative stress that characterises the syndrome.
Therefore, the expression of ferritin and transferrin receptors (TfR) were sought in the lungs and hearts of rodents treated with lipopolysaccharide (LPS), and measurements of TfR and ferritin protein expression were taken from lung biopsy specimens from patients with ARDS and appropriate controls.
TfR messenger ribonucleic acid (mRNA) was significantly upregulated in the lungs and significantly downregulated in the hearts of rats 4 h after LPS. Ferritin mRNA levels (light and heavy chains) remained unaltered. Protein TfR levels were significantly upregulated in lungs and downregulated in hearts 4 h post-LPS. Ferritin protein levels were significantly downregulated in lungs compared to baseline values but were unaltered in hearts. Nonhaem iron levels were increased in lungs and decreased in hearts, and iron-regulatory-protein activity increased in hearts but not lungs. TfR protein levels were significantly increased in lung biopsies from patients with ARDS compared to controls.
Transferrin receptors are upregulated in rodent lungs during inflammation but are downregulated in the heart. Transferrin receptor protein levels were significantly increased in the lungs in clinical acute respiratory distress syndrome. These findings have implications for the pathogenesis of acute respiratory distress syndrome, especially in relation to the role of iron as a mediator of oxidative stress.
This study was supported by a British Lung Foundation/BOC Group Programme Grant, the Dunhill Medical Trust and British Heart Foundation. ¶Joint first authors.
Acute respiratory distress syndrome (ARDS) in adults is defined clinically by refractory hypoxaemia and bilateral pulmonary infiltrates, attributable to increased permeability of the alveolar capillary membrane. ARDS may complicate a wide variety of infective and noninfective pro-inflammatory insults, not all of which involve the lung directly. Why such provocative factors should render the lung, rather than other organ systems, susceptible to the inflammatory processes implicated in the pathogenesis of the syndrome is, however, unclear 1. Patients with ARDS are also subjected to conditions of severe oxidative stress, which are implicated in the pathogenesis of the inflammatory processes that characterise the condition. Thus, abnormalities in plasma iron chemistry (elevated levels of iron saturation of transferrin) and impaired antioxidant protection (normally afforded by the binding of iron to transferrin and the iron-oxidising, ferroxidase activity of caeruloplasmin) have been described previously 2, and bronchoalveolar lavage fluid from patients with ARDS has been shown to contain increased levels of low molecular mass iron compared to that taken from normal healthy controls 3. Iron is a catalyst at the active centres of numerous oxidase, oxygenase and antioxidant enzymes, and in other proteins for the transport of electrons and oxygen, but when not under such protein structural constraints it is capable of catalysing the formation of numerous aggressive and damaging inorganic and organic reactive oxygen species (ROS) 4, 5.
To avoid the deleterious consequences of iron misuse, elaborate physiological mechanisms have evolved to ensure its efficient recycling, turnover and control 6. These include the expression of transferrin receptors (TfRs) on cells to facilitate the uptake of iron-loaded transferrin and the expression of cellular apoferritin (both heavy and light chains) in which to store iron intracellularly. The expression of these two proteins is regulated at the post-transcriptional level by the iron-regulatory proteins (IRP)‐1 and ‐2. Activated IRP‐1 and nondegraded IRP‐2 bind to iron-responsive elements (IRE) in the noncoding regions of ferritin and TfR messenger ribonucleic acid (mRNA). Such responses are controlled by the prevailing levels of iron (albeit by different mechanisms for IRP‐1 and ‐2). When cellular iron levels are low, inhibition of ferritin synthesis occurs together with a stabilisation of TfR mRNA leading to increased TfR synthesis. By contrast, when iron levels are high the reverse process occurs 7, 8. Mechanisms of IRP activation independent of iron signalling have also been reported to contribute to iron regulation. These include activation by ROS 9 and inflammatory mediators 10.
Therefore, the presence of iron and its regulation has a potentially crucial part to play in the pathogenesis of ARDS. However, the extent to which dysfunctional iron regulation might render the lung particularly susceptible to injury following pro-inflammatory insults has not been explored. The aims of this study were, therefore, three-fold. First, usinga rodent model, the effects of a pro-inflammatory insult (lipopolysaccharide (LPS)) on the mobilisation of iron (cellular uptake and storage) were investigated through the expression of TfRs and ferritin. Secondly, the hypothesis thatthe expression of these iron uptake and storage proteinswas organ-, and by inference, cell-specific was explored, since differences may have important implications for the pathogenesis of ARDS. Finally, evidence of TfR and ferritinexpression was sought in archived lung biopsy specimens taken from patients with ARDS and appropriate controls.
Materials and methods
Animal studies
All procedures and protocols were performed in accordance with the Animal (Scientific Procedures) Act, 1986, and approved by the Home Office (UK) Inspectorate. Male Wistar rats (n=4 per group except where otherwise stated, 275–300 g) received LPS (20 mg·kg−1 i.p.) or an equivalent volume of saline. Either 120 or 240 min post-LPS, the animal was anaesthetised with pentobarbitone (60 mg·kg−1 i.p.), sacrificed by cervical dislocation, and the heart and lungs were harvested for reverse transcriptase polymerase chain reaction (RT‐PCR) and Western blotting.
Clinical studies
Archived lung tissue biopsies (stored at −70°C) taken for diagnostic purposes from patients with ARDS (n=7) and defined by America-European Consensus Conference criteria (for patient demographics see table 1⇓), and normal tissue from seven control patients (six male and one female) undergoing lung resection for bronchogenic or metatastic carcinoma (mean age 58 yrs, range 45–75 yrs), were homogenised and processed. TfR and ferritin protein levels were determined using electrophoresis and protein blotting. The methodology employed was as for the animal studies described below.
Demographic details fulfilling American-European Consensus criteria for acute respiratory distress syndrome (ARDS)
Reverse transcription and polymerase chain reaction
Rodent tissues were homogenised and ribonucleic acid (RNA) was extracted using Tri Reagent (Sigma-Aldrich Company Ltd, Poole, UK). Single stranded complementary deoxyribonucleic acid (cDNA) was synthesised with the aid of Moloney murine leukemia virus reverse transcriptase (InvitrogenLtd, Paisley, UK) and oligo (dT)12–18 primers (Invitrogen Ltd). Oligo primers were chosen as described in previous studies for the transferrin receptor 11, ferritin-heavy 12 and -light chains 13, and for IRP‐1 and ‐2 14, 15. Double stranded cDNAs were synthesised and amplified by use of Taq deoxyribonucleic acid (DNA) polymerase (Invitrogen Ltd). A total of 27 cycles were performed for TfRs and 17 cycles for both ferritin-heavy and -light chains. Semi-quantitative analyses were performed with reference to β‐actin (16 cycles performed), which was selected for use as a housekeeping gene. After gel electrophoresis, densitometric analysis was performed with National Institute of Health Image software (linear response over four optical density units).
Western blotting
Rodent hearts and lungs, and human lung tissue were homogenised with a pH 7.0 cell lysis buffer containing proteinase inhibitor CompleteTM (Roche Diagnostics, Lewes, UK), and centrifuged at 10,000×g for 10 min. Supernatants were divided into aliquots and total protein determined by the Lowry method. Volume adjustments weremade with loading buffer to ensure that samples containing equal amounts of protein were used for electrophoresis. The protein concentration used for a given analysiswas established by trial and error, and differed according to the type and tissue. For TfR studies in hearts,wells were loaded with 25 µL of 1.0 µg·µL−1, and for those in lungs, 2.0 µg·µL−1. For ferritin studies in hearts, wellswere loaded with 25 µL of 0.2 µg·µL−1, and in lungs,0.4 µg·µL−1. In heart-derived samples, TfR gel electrophoresis was performed under nondenaturing conditions (7.5% weight/volume (w/v) gel), which facilitated the movement and determination of the parent TfR homodimer. Inlung supernatants, it was decided for technical reasons to perform TfR electrophoresis using standard denaturing (inthe presence of sodium dodecylsulphate (SDS) and after heating) conditions (15% w/v gel). In this way the subunit corresponding to the TfR monomer was separated and determined. Ferritin electrophoresis was also performed under standard denaturing conditions because of the largesize of the parent multi-subunit protein and again with 15% w/v SDS‐PAGE gels.
Gels were transferred onto nitrocellulose membranes. Afterblocking, blots were probed with a polyclonal antibodyagainst TfR (Bioquote Ltd, York, UK) or ferritin(Sigma-Aldrich Ltd). The samples were then incubated with a second anti-immunoglobulin G horseradish peroxidase conjugate. Membranes were treated with enhanced chemiluminescence reagent to reveal immunoreactive proteins. A TfR or ferritin standard was run on each gel as an added control. Results are expressed as mean densitometric readings.
Total nonhaem iron measurement
These were measured in tissue homogenates prepared in the absence of iron chelators using iron-free reagents. A kit method was employed (Total Iron Assay Kit; Sigma Chemical Company, Dorset, UK).
Bleomycin-detectable iron measurement
Free iron can be chelated by bleomycin, which is the basis of this assay. The protocol is fully described in 16.
Iron-regulatory protein activity
IRP activity was determined by use of an RNA gel shift assay technique, as described previously 17. A vector plasmid (pTZ19‐IRE) containing the IRE for heavy-chain ferritin was obtained from Fermentas AB (Vilnius LT, Lithuania). The plasmid was linearised and 32P labelled RNA was transcribed to produce the radiolabelled IRE probe. Protein extracts (2 µg) from hearts and lungs were mixed with saturating amounts of the IRE probe (1.3×105), and incubated at ambient temperature for 10 min. Heparin displaced nonspecific interactions. β‐Mercaptoethanol (2%) was added to one set of samples for total IRP activation; for endogenous activity β‐mercaptoethanol was omitted. RNA‐protein complexes were resolved on native 6% polyacrylamide gels. Gels were developed by autoradiography. In work-up experiments with smooth muscle cells, IRP‐1 and ‐2 activity could be differentiated. However, IRP‐2 activity could not be consistently demonstrated in samples derived from the in vivo model. This is probably due to the known labile nature of IRP‐2, the activity of which may be lost on processing tissue samples to release protein.
Statistical analysis
Results are expressed throughout as mean±sem. Where two groups were compared, differences were assessed using the Mann-Whitney U‐test. One way analysis of variance (ANOVA) and Dunnett's multiple comparisons test were used for comparisons of multiple groups. A p‐value of <0.05 was considered significant.
Results
Rats treated with LPS expressed significantly decreased levels of TfR mRNA in the heart over time. Thus, in the heart, differences between groups were significant overall (p=0.036). Post-tests revealed a significant downregulation of TfR mRNA expression 4 h after LPS, as compared to time 0 (fig. 1a⇓). Decreased levels of TfR protein were also observed. Although these differences were not significantly different overall, the trend was for a decrease (fig. 1b⇓). However, one data point in the control group was skewed; with this point removed, differences reached overall significance (p=0.016). Furthermore, post-tests showed a significant decrease for TfR protein 4 h after LPS, as compared to time 0.
Levels of transferrin receptor (TfR) in the heart of rats. a) Levels of expression of TfR messenger ribonucleic acid (mRNA), measured as densitometric ratios relative to the β‐actin housekeeping gene (n=4 for all groups) and a representative blot for TfR and β‐actin mRNA after reverse transcriptase polymerase chain reaction (RT‐PCR) amplification. b) Levels of expression of TfR protein measured as denisitometric values (n=4 for all groups) and a representative Western blot of TfR protein from the heart. Levels are shown both pre- (□; 0 h) and post- (└; 2 and 4 h) lipopolysaccharide treatment. *: p<0.05.
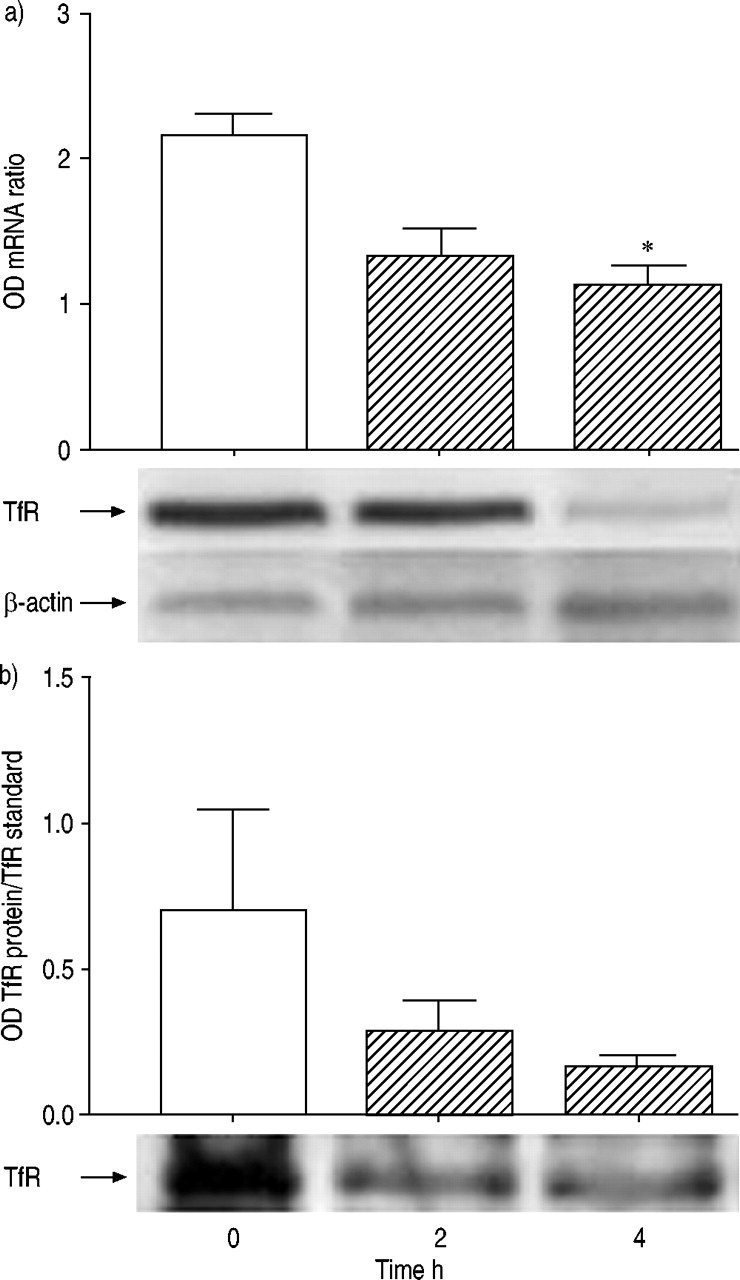
In the lungs from rats treated with LPS the opposite response was observed. Thus, TfR mRNA levels increased over time. Differences between groups were significant overall (p=0.04). Post-tests revealed a significant upregulation of TfR mRNA at 4 h after LPS, as compared with time 0 (fig. 2a⇓). Comparable increases in TfR protein were also seen in the lungs. Differences between groups were significant overall (p=0.0066). An obvious and significant increase in TfR protein was apparent 4 h (n=5) after LPS, as compared to baseline (n=6) (fig. 2b⇓), reflecting the changes observed in mRNA levels at the same time point.

Levels of transferrin receptor (TfR) in the lungs of rats. a) Levels of expression of TfR messenger ribonucleic acid (mRNA), measured as densitometric ratios relative to the β‐actin housekeeping gene (n=4 for all groups) and a representative blot for TfR and β‐actin mRNA after reverse transcriptase polymerase chain reaction (RT‐PCR) amplification. b) Levels of expression of TfR protein, measured as denisitometric values (n=4 for all groups) and a representative Western blot of TfR protein in the lungs. Levels are shown both pre- (□; 0 h) and post- (└; 2 and 4 h) lipopolysaccharide treatment. *: p<0.05.
No significant alterations in the expression of ferritin light- or heavy-chain mRNA were observed overall or at any time point in either rodent heart or lungs (data not shown).
Ferritin protein levels in the heart remained unaltered at all time points (fig. 3a⇓). In the lungs, differences between groups were significant overall (p=0.031). Post-tests showed a significant downregulation of ferritin protein expression in thelungs 4 h after LPS, as compared to baseline (fig. 3b andc⇓). Changes in mRNA levels do not exclude a post-transcriptional control mechanism.

Levels of expression of ferritin protein, measured as densitometric ratios relative to a ferritin protein standard, in a) the heart and b) the lungs of rats (n=4 in all cases). c) A representative Western blot showing ferritin protein (heavy chain) in the lung. Levels are shown both pre- (□; 0 h) and post- (└; 2 and 4 h) lipopolysaccharide treatment. *: p<0.05.
IRP‐2 mRNA levels remained unaltered in both the heart and lungs after LPS challenge (data not shown). However, IRP‐1 mRNA levels were clearly but not quite statistically significantly (p=0.057) reduced in the heart 2 and 4 h after LPS. No such changes were apparent in the lungs.
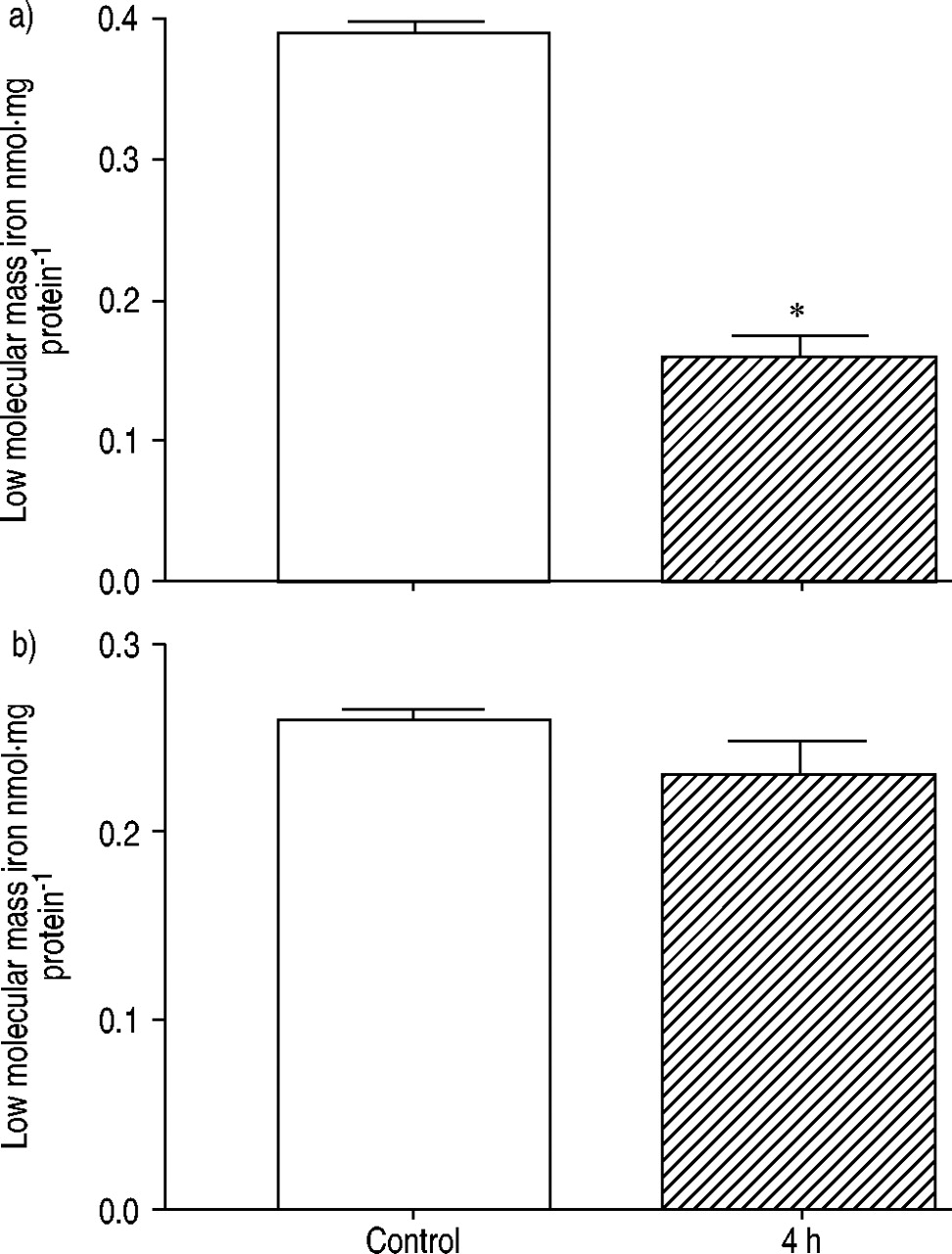
Levels of total nonhaem iron were measured at baseline and at 4 h in control (n=6) and LPS‐treated (n=3) rats. An increase was seen in the lungs (fig. 4a⇓), but decreases in total nonhaem iron 4 h post‐LPS, as compared to baseline values, were seen in the heart (fig. 4b⇓). Interestingly, levels of low molecular mass/loosely bound (bleomycin-detectable) iron were significantly (p<0.05) decreased in the lungs 4 h after LPS, as compared to baseline (fig. 5a⇓). By contrast, in the heart, the pattern mirrored those seen for total nonhaem iron (albeit at lower iron concentrations) (fig. 5b⇓).

Changes in total nonhaem iron in a) lung and b) heart homogenates from control animals (□; n=6) and 4 h post-administration of lipopolysaccharide (└; n=3).
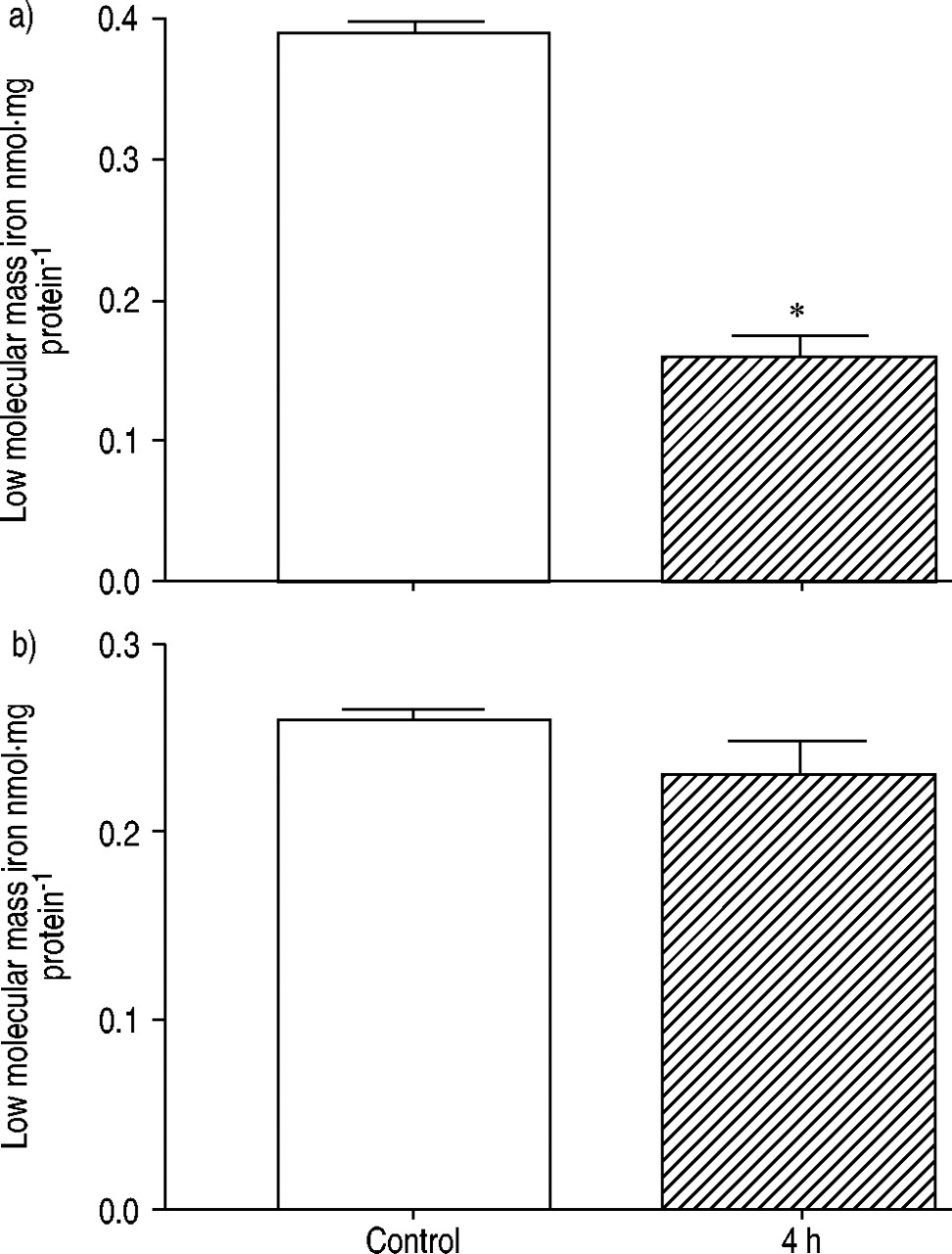
Changes in bleomycin-detectable iron (low molecular mass iron) in a) lung and b) heart homogenates from control animals (□; n=6) and 4 h post-administration of lipopolysaccharide (└; n=3). *: p<0.05.
IRP activity could be demonstrated in all samples (n=3 for all groups) in which it was measured (fig. 6a and b⇓). There was a significant increase in IRP activity in the hearts from LPS‐treated animals after 4 h, as compared with baseline levels (fig. 6a⇓), but no change was demonstrated in the lungs (fig. 6b⇓).

Levels of iron-regulatory protein (IRP) activity (n=3 for both groups), measured as densitometric ratios relative to total activity in a) the heart and b) lungs of control (□) and lipopolysaccharide-treated (└) rats. *: p<0.05.
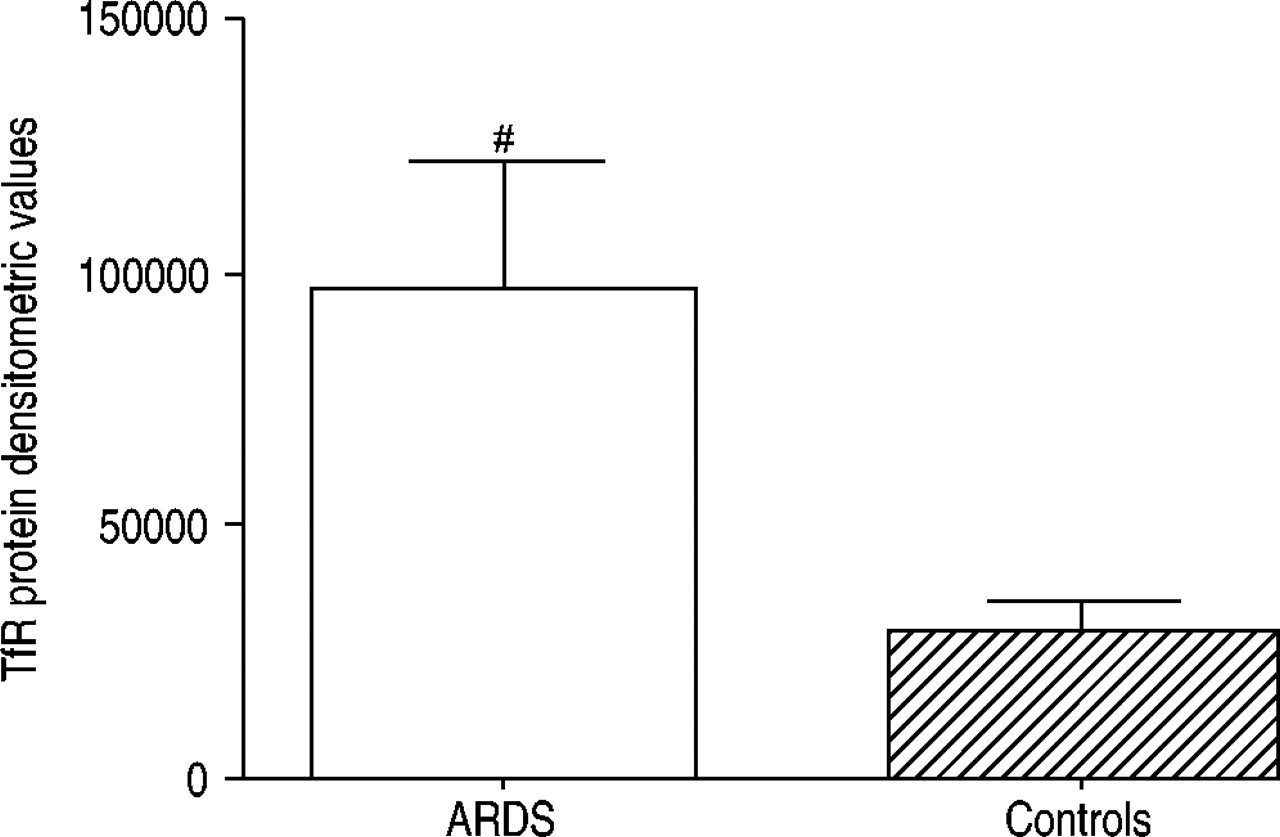
Significantly increased (p=0.02) levels of TfR protein were found in lung tissue taken from patients with ARDS (n=7), as compared to controls (n=7) (fig. 7⇓). However, there were no obvious differences in ferritin protein levels between groups (3854±1196 versus 4255±5410, p>0.05). It was not possible to measure mRNA levels as the samples had been stored for some time.

Transferrin receptor (TfR) protein levels in lung tissue homogenates from patients with acute respiratory distress syndrome (ARDS) (n=7) and appropriate controls (n=7). Equivalent concentrations of total protein were used throughout. Levels of TfR were significantly increased (#: p=0.02) in patients with ARDS.
Discussion
This investigation indicates that a pro-inflammatory insult in an experimental model results in preferential upregulation of TfRs in the lungs, with a concomitant downregulation in the heart. TfR protein levels were significantly increased in the lungs in clinical ARDS. These findings may explain, in part, the apparent susceptibility of lung tissue to inflammatory insults and may also have implications for the pathogenesis of ARDS, especially in relation to the role of iron as a mediator of oxidative stress.
In endotoxaemia, increased levels of TfR mRNA and protein were observed in lung tissue when compared to controls, suggesting a degree of transcriptional regulation of the expression of this protein, although increased mRNA stability cannot be conclusively excluded as contributing to this finding. However, the four-fold increase in lung TfR mRNA at the early (2 h) time point strengthens the argument for LPS‐mediated transcriptional effects. Ferritin was significantly decreased in lung tissue, whilst the level of mRNA for both subunits remained unaltered. This suggests that regulatory control at the translational level occurred. However, there was no apparent change in IRP activity in the lungs. This discrepancy is difficult to explain, but if correction for total activity is not applied, apparent changes in activity can be seen (fig. 8a⇓, lanes 1 and 2).

Representative ribonucleic acid (RNA) gel shift assay showing iron regulatory protein (IRP)‐1 activity in a) the lungs and b) theheart pre-treatment (1 and 3) and 4 h post-lipopolysaccharide treatment (2 and 4). The total activities were obtained by adding 2‐mecaptoethanol (3 and 4).
The changes observed in TfR message and protein values in heart tissue were the opposite of those seen in the lung. As in the heart, levels of mRNA and protein were significantly decreased. Ferritin message and protein levels did not appear to change in the heart. Regulation of TfR and ferritin expression is viewed as a post-transcriptional event mediated by IRP‐1 and ‐2, although transcriptional regulation also occurs 6, 18. Therefore, IRP activity was examined in heart and lung tissues obtained from LPS‐treated rodents; however, IRP‐2 activity could not be measured, most probably because IRP‐2 activity is lost during tissue processing. Nevertheless, measurable IRP activity was significantly upregulated in the heart. This result is somewhat at variance with the current authors' finding of unaltered ferritin and decreased TfR expression, since a downregulation of IRP activity would be expected in these circumstances. Interestingly, IRP‐1 mRNA levels decreased in heart tissue after LPS, a finding that may go some way to explaining this discrepancy. The failure of LPS to elicit a change in IRP activity in the lungs when normalised to total activity, even though there were pronounced changes in the expression of TfRs and ferritin, is also somewhat puzzling. However, when uncorrected endogenous activity is compared (fig. 8⇑, lanes 1 and 2), IRP activity was increased. Therefore, the possibility that LPS increases IRP activity in the lung cannot be excluded, although it does seem likely that a transcriptional regulatory component for TfR expression is involved.
Several studies have demonstrated decreased expression of TfR in response to pro-inflammatory stimuli such as LPS in combination with γ‐interferon, tumour necrosis factor, interleukin‐1 and combinations of chemokines applied to isolated epithelial cells and or macrophages 19–21. Indeed, nitric oxide-mediated degradation of IRP‐2 is a key regulatory component under such circumstances 22, 23. As macrophages and epithelial cells are found in the lungs, these studies may be of relevance, however, the present in vivo studies oppose those above in that an increase in TfR levels was seen in the lungs. Hypoxia inducible factor‐1‐mediated transcriptional regulation of TfR expression in response to hypoxia has also been demonstrated 18. Hypoxia may be highly relevant to this in vivo study as a defining criterion for ARDS. The current authors', as yet, unpublished studies using the same in vivo model (data not shown) are investigating the relationship between haemoxygenase-mediated iron release and the modulation of IRP activity in vivo. Their preliminary findings suggest that iron is a key regulatory component for the modulation of measurable IRP activity in the lungs, but that other mechanisms, possibly related to oxidative stress, are more important in the heart.
In functional terms, the current data indicate that following LPS, iron uptake and storage mechanisms are regulated in such a way as to favour decreased cellular uptake of iron by the rodent heart. In the lungs, the opposite appears to occur (increased TfR and decreased ferritin expression), a situation that would favour iron accumulation. Indeed, preliminary studies suggest that 4 h after LPS, tissue nonhaem iron levels are decreased in the heart but increased in the lungs. Although the lungs may therefore accumulate nonhaem iron in sepsis, mechanisms appear to exist to limit any adverse effects associated with the cellular accumulation of low molecular mass forms of iron, since levels of bleomycin-detectable iron decreased. The storage of iron by ferritin could, in part, account for these findings, although this protein was found to be decreased in the lung in response to LPS in the current study. Other iron-binding mechanisms may account for the sequestration observed, including that of the intracellular iron transport protein Nramp1.
The current authors suggest that the opposing responses demonstrated here, as summarised in figure 9⇓, are related to organ/cell-type specific adaptations that increase antioxidant and antimicrobial protection against iron. Specifically, they propose that the heart, by decreasing TfR expression, may protect itself against the potential pro-oxidant effects of iron. This could be particularly important during acute inflammatory conditions, such as those encountered in this model, since this is a time of increased ROS production 24. The high respiratory demand of the heart may also make it more susceptible to oxidative damage in the presence of redox active iron. Moreover, iron is known to be acutely cardiotoxic 25. Such a response is consistent with an antioxidant response evolved to protect the heart from oxidative damage.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Schematic diagram summarising the changes in iron-regulatory responses observed in the heart and lungs in response to alipopolysaccharide (LPS) challenge. +−: represents no change; −−: decrease; ++: increase. IRP: iron regulatory protein; mRNA: messenger ribonucleic acid.
However, the increased expression of TfRs and probable iron uptake by the lungs is more difficult to interpret, since iron is potentially both pro-oxidant and promotes microbial growth. The balance between these two consequences may be critical in the development of ARDS in patients with sepsis. For example, iron may normally be used as part of a rapid pro-oxidant response to kill bacteria in the lung, pre-empting its ability to promote microbial virulence. ARDS is an acute inflammatory condition complicating a diverse array of predisposing insults 26. Sepsis remains the most common precipitating condition for ARDS 27, 28. Oxidative stress, leading to molecular damage and abnormal iron chemistry, has been identified as an important factor contributing to morbidity and mortality 2. In patients with sepsis, the lung is usually the first organ to be compromised, but the associated systemic inflammatory response means that all organs can be adversely affected 29. The current findings (fig. 9⇑) provide the potential for considerable oxidative stress during an acute inflammatory response. Indeed, iron uptake by macrophages has been shown to increase cellular injury under conditions of hyperoxia 30. The changes in iron regulation observed during endotoxaemia may offer an explanation as to why the lung appears to be so vulnerable in clinical sepsis. Findings of increased TfR protein levels in lung tissue samples obtained from patients with ARDS, when compared to appropriate controls, corroborates the findings from the in vivo model, although ferritin levels were not different between groups.
In conclusion, the responses observed in this study are likely to represent adaptations to increase antioxidant and antimicrobial protection against iron. The presence of extracellular iron in normal healthy lungs 31 makes it particularly vulnerable to oxidative damage and to infection when such control is lost.
- Received August 15, 2002.
- Accepted March 28, 2003.
- © ERS Journals Ltd
References









