





Abstract
Inhibition of Rho-associated coiled-coil forming kinases (ROCKs) reduces allergic airway responses in mice. The purpose of this study was to determine the roles of the two ROCK isoforms, ROCK1 and ROCK2, in these responses.
Wildtype (WT) mice and heterozygous ROCK1 and ROCK2 knockout mice (ROCK1+/- and ROCK2+/-, respectively) were sensitised and challenged with ovalbumin. ROCK expression and activation were assessed by western blotting. Airway responsiveness was measured by forced oscillation. Bronchoalveolar lavage was performed and the lungs were fixed for histological assessment.
Compared with WT mice, ROCK1 and ROCK2 expression were 50% lower in lungs of ROCK1+/- and ROCK2+/- mice, respectively, without changes in the other isoform. In WT lungs, ROCK activation increased after ovalbumin challenge and was sustained for several hours. This activation was reduced in ROCK1+/- and ROCK2+/- lungs. Airway responsiveness was comparable in WT, ROCK1+/-, and ROCK2+/- mice challenged with PBS. Ovalbumin challenge caused airway hyperresponsiveness in WT, but not ROCK1+/- or ROCK2+/- mice. Lavage eosinophils and goblet cell hyperplasia were significantly reduced in ovalbumin-challenged ROCK1+/- and ROCK2+/- versus WT mice. Ovalbumin-induced changes in lavage interleukin-13, interleukin-5 and lymphocytes were also reduced in ROCK1+/- mice.
In conclusion, both ROCK1 and ROCK2 are important in regulating allergic airway responses.
Asthma is characterised by bronchoconstriction, airway hyperresponsiveness (AHR) and airway infiltration with T-lymphocytes and eosinophils. While the pathogenesis of asthma has not been fully established, evidence from animal models suggests that Rho-associated coiled-coil forming kinases (ROCKs) may be involved. ROCKs are serine/threonine protein kinases that are activated by guanosine triphosphate (GTP)-bound RhoA. ROCKs target many substrates and have been shown to have diverse cellular functions, including smooth muscle contraction, stress fibre formation, cell migration and proliferation [1, 2]. GTP binding to RhoA is regulated by guanine nucleotide exchange factors (GEFs), which are activated following ligand binding to many tyrosine kinase and G protein-coupled receptors, including many of the mediators of allergic asthma [3]. In addition, RhoA protein expression is increased in bronchi of allergen-sensitised and -challenged rats and guinea pigs [4, 5]. Finally, in ovalbumin (OVA)-sensitised and -challenged guinea pigs and mice, the ROCK inhibitors, Y-27632 and fasudil, suppress AHR and attenuate eosinophilic airway inflammation and expression of T-helper cell (Th) type 2 cytokines [6–9]. However, there are issues of specificity with these agents, since they can also inhibit other protein kinases [10, 11].
Two ROCK isoforms have been identified: ROCK1 and ROCK2 [1, 12]. They share 92% homology in their kinase domains, suggesting that they target the same amino acid motifs. However, their Rho-binding domains are only 55% homologous, and there is evidence that they can be differently regulated [13–16]. There are also tissue-specific differences in ROCK1 and ROCK2 expression: ROCK1 mRNA is abundant in liver, stomach, spleen and kidney, whereas ROCK2 expression is high in brain and muscle [12]. The subcellular localisation of ROCK1 and ROCK2 is also different: ROCK1 is expressed diffusely and perinuclearly, while ROCK2 is expressed both perinuclearly and at cell margins [17]. Taken together, these data indicate that ROCK1 and ROCK2 may mediate different functions in different cell types.
The purpose of this study was to examine the specific contributions of ROCK1 and ROCK2 to allergic airways disease in mice. Available pharmacological inhibitors of ROCK target the ATP-dependent kinase domains of ROCK1 and ROCK2 and are thus not isoform specific. Consequently, we used ROCK1- and ROCK2-deficient mice to address this issue. Most homozygous ROCK1- or ROCK2-deficient mice die either in gestation or early postnatally [18, 19]. However, heterozygous ROCK knockout mice (ROCK1+/- and ROCK2+/-) are viable, and express only about 50% of wildtype (WT) levels of the affected ROCK isoform without compensatory changes in the other isoform [20, 21]. Accordingly, we assessed pulmonary responses of ROCK1+/- and ROCK2+/- mice to OVA sensitisation and challenge as previously described [22]. We also assessed ROCK activation following OVA challenge using western blotting to measure phosphorylation of the myosin-binding subunit (MBS) of myosin light chain phosphatase (MLCP), as previously described [23, 24]. MBS is a specific ROCK substrate [23].
METHODS
Animals
This study was approved by the Harvard Medical Area Standing Committee on Animals. Mice were generated as previously described [20, 21]. Because the ROCK1+/- and ROCK2+/- knockout mice were on a C57BL/6 background, age- and sex-matched WT C57BL/6 mice were used as controls. Approximately half the WT mice were littermate controls of the ROCK1+/- or ROCK2+/- mice and the other half were purchased from Jackson Labs (Bar Harbor, ME, USA). We observed no difference between the littermate and purchased mice for any of the outcome indicators measured.
Allergen sensitisation and challenge
4-week-old male mice were sensitised to chicken egg albumin (Grade V; Sigma-Aldrich Co., St Louis, MO, USA) on day 0 by i.p. injection of 20 μg of OVA and adjuvant, 2 mg of aluminium hydroxide (Al(OH)3; J.T. Baker, Phillipsburg, NJ, USA) dispersed in 0.2 mL of PBS. The mice were given a second injection of identical reagents on day 14. On days 28–30, mice were challenged for 30 min with an aerosol of either PBS containing 1% OVA or PBS alone, as previously described [25].
Assessment of ROCK expression and activation
At several time-points after the last challenge, mice were euthanised and the lungs quickly harvested and frozen in liquid nitrogen. To assess ROCK activation, lung homogenates were subsequently used for western blotting with an antibody to phospho-Thr853 MBS as previously described [23]. Thr853 is a specific substrate for ROCK [23]. Rabbit anti-MBS polyclonal antibody (Covance Laboratories, Indianapolis, IN, USA) was used to detect the total amount of MBS [23]. NIH 3T3 cell lysates were used as a positive control and to standardise the results of western blot analyses. Pulmonary expression of ROCK1 and ROCK2 were measured by western blot with anti-ROCK2 monoclonal antibody, anti-ROCK1 monoclonal antibody (BD Transduction Laboratories, San Jose, CA, USA) or anti-actin monoclonal antibody (Sigma-Aldrich).
Measurement of pulmonary mechanics and airway responsiveness
Mice were anaesthetised with xylazine (7 mg·kg−1) and sodium pentobarbital (50 mg·kg−1) 48 h after the last OVA or PBS challenge and instrumented for the measurement of pulmonary mechanics and airway responsiveness to i.v. methacholine using the forced oscillation technique, as previously described [22]. In naïve WT mice, we also assessed the effects of the combined ROCK1 and ROCK2 inhibitor, fasudil [26], on airway responsiveness to inhaled methacholine. Fasudil (10 mg·kg−1 i.p.) was administered 30 min prior to measurement of mechanics. Mice that served as controls for fasudil-treated mice received an equal volume of the vehicle used to dissolve fasudil, which was PBS. This dose of fasudil has been shown by others to cause a ∼50% inhibition of ROCK activation in ischaemic regions of the brains of mice subjected to middle cerebral artery occlusion [27]. For these measurements, methacholine was administered as 10 breaths of an aerosol derived from concentrations of methacholine of 0.3–100 mg·mL−1. Pulmonary resistance (RL) was measured every 10 s for 2 min after aerosol administration. In each mouse, at each dose, the three highest RL values were averaged to obtain a final value.
Bronchoalveolar lavage
Bronchoalveolar lavage (BAL) was performed and the BAL cells and differentials counted as described previously [22, 25]. BAL supernatant was stored at -80°C and subsequently analysed by ELISA for interleukin (IL)-13 and IL-5 (R&D Systems Inc., Minneapolis, MN, USA).
Histology and morphometry
Lungs were removed from mice and inflated with 6.25% glutaraldehyde or 10% formalin to a pressure of 23 cmH2O. After fixation, the left lung was sectioned and embedded in paraffin. Mucus-containing goblet cells were stained using periodic acid–Schiff stain. Peripheral airways were viewed using an upright light microscope equipped with SPOT CCD camera and ImagePro software (Olympus Canada, Mississauga, ON, Canada). Specific airways were selected for analysis using the criteria described by Hirota et al. [28]. Basement membrane (BM) length was measured for all airways. For measurements, a band-shaped region of interest extending from the BM to the edge of the epithelium was drawn. For all analyses, images of at least three different airways were captured from three or four sections for three different animals in each group. Other sections were stained with haematoxylin and eosin to determine the inflammation index, a product of the severity and prevalence of inflammation, as described by Hamada et al. [29]. Severity was assigned a numerical value based upon the number of inflammatory cell infiltrate layers around the airways or blood vessels (0: no cells; 1: ≤3 cell layers; 2: 4–9 cell layers; 3: ≥10 cell layers). The prevalence of inflammation was assigned a numerical value according to the percentage of airways or blood vessels in each section encompassed by inflammatory cells (0: no airways or vessels; 1: ≤25%; 2: 25–50%; 3: >50%). Medium-to-large sized airways and vessels were analysed and, in each mouse, a minimum of eight airways and eight vessels were examined. A mean value for each mouse was computed and used for statistical analyses.
Dendritic cell/T-lymphocyte co-cultures
To determine whether ROCK might be affecting dendritic cell activation of T-lymphocytes, spleens were harvested from DO.11 mice (Jackson Labs). These transgenic mice express the mouse α-chain and β-chain T-cell receptor that pairs with the CD4 co-receptor and is specific for chicken OVA 323–339 peptide in the context of major histocompatibility complex class II. In these mice, T-cells demonstrate a dose-dependent proliferative response to the specific OVA ligand. T-cells from DO.11 mice and dendritic cells from Balb/c mice were isolated from splenocyte suspensions as described [30]. Dendritic cells and T-cells were then co-cultured (0.25×105 dendritic cells with 1.2×105 lymphocytes) in 100 μL of RPMI complemented with fetal bovine serum, l-glutamine and penicillin–streptomycin in 96-well plates. Once plated, OVA protein (Sigma-Aldrich) was added to stimulate dendritic cell presentation of OVA to the T-cells; control wells were left unstimulated. To assess lymphocyte proliferation in response to OVA, tritiated thymidine (Sigma-Aldrich) was added. 48 h later, cells were washed to remove unincorporated thymidine and then lysed and the radioactivity counted. In some wells, the ROCK inhibitor Y-27632 [31] was added prior to OVA stimulation. Control wells were treated with the vehicle used to dissolve Y-27632, which was the cell growth medium.
Statistical analysis of results
Data were analysed by factorial ANOVA using STATISTICA software (StatSoft®; Tulsa, OK, USA). Fisher's least significant difference test was used for post hoc comparisons. A p-value <0.05 was considered statistically significant.
RESULTS
Time-course of OVA-induced ROCK activation
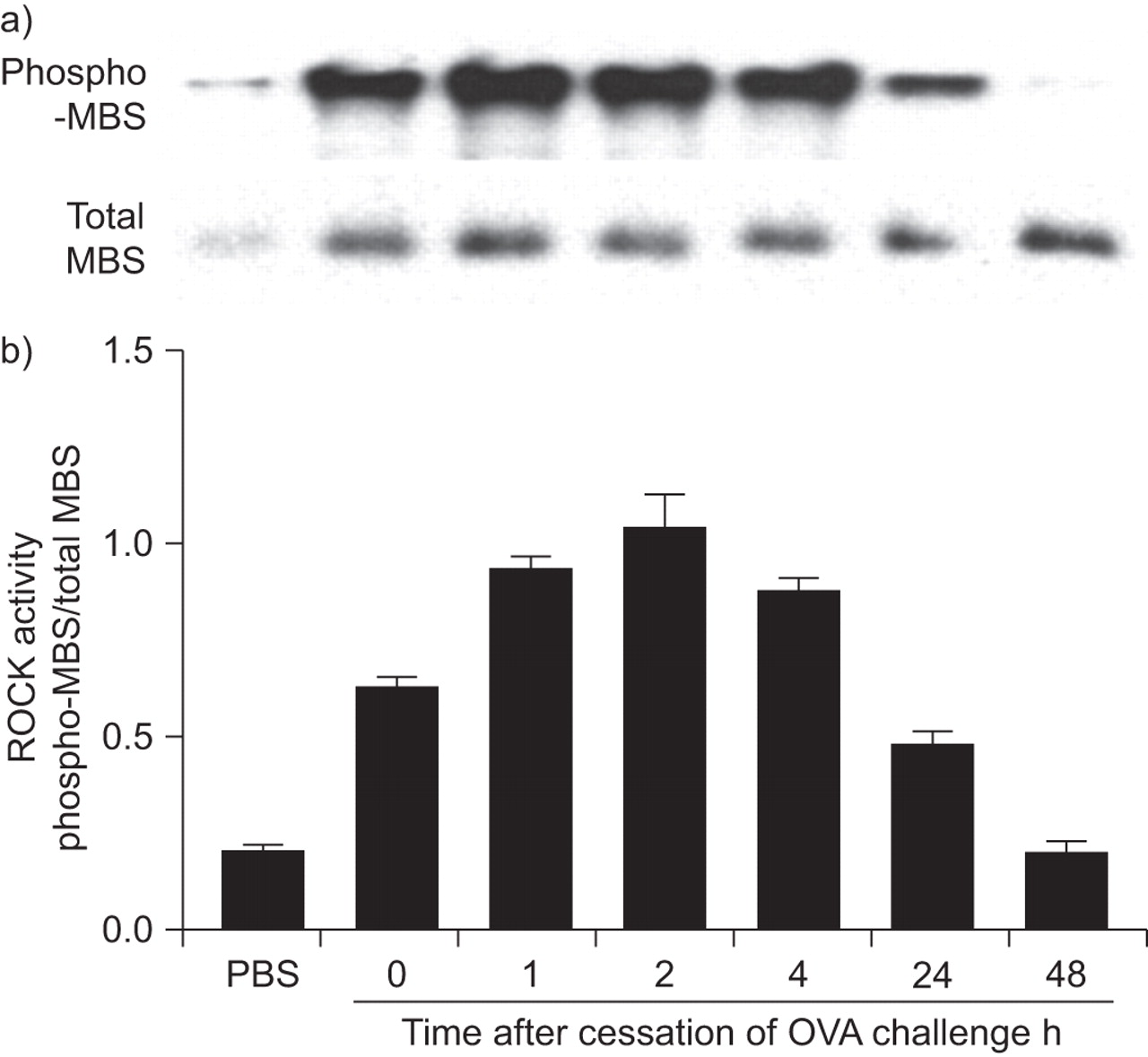
Western blotting for phospho-MBS, a specific indicator of ROCK activation [23], was used to assess ROCK activation. OVA challenge resulted in an immediate increase in ROCK activity (fig. 1). Compared with challenge with aerosolised PBS, phospho-MBS staining (normalised for total MBS) was significantly increased in mice euthanised immediately after removal from the OVA exposure chamber (fig. 1; 0 h) and was sustained for at least 4 h after OVA exposure before beginning to decline. Note that there was some constitutive ROCK activation in the lungs even after PBS challenge.

Time-course of ovalbumin (OVA)-induced Rho-associated coiled-coil forming kinase (ROCK) activation. a) Western blots showing phosphorylated (phospho)-myosin-binding subunit (MBS) or total MBS in protein isolated from wildtype mice sensitised to OVA and challenged on three consecutive days with aerosolised OVA or PBS. Lungs were harvested immediately (0 h) or 1, 2, 4, 24 or 48 h after the last OVA aerosol challenge. b) Densitometric analysis showing mean±se of data from three mice in each group. ROCK activity was expressed as the percentage of phospho-Thr853-MBS in each sample or positive control divided by total MBS in each sample or positive control.
ROCK1 and ROCK2 expression
Western blotting was used to confirm reductions in pulmonary expression of ROCK1 and ROCK2 in ROCK1+/- and ROCK2+/- mice (fig. 2). Densitometric analysis confirmed a ∼50% reduction of ROCK1 in ROCK1+/- mice and a similar reduction of ROCK2 in ROCK2+/- mice without compensatory changes in the other isoform. Changes in ROCK expression in these mice resulted in reduced ROCK activity: compared with WT mice, phospho-MBS was significantly reduced in lung tissue of ROCK1+/- and ROCK2+/- mice studied 48 h after the last exposure to PBS or ovalbumin (fig. 3). The reduction in ROCK activation was particularly pronounced in ROCK2+/- mice, in which phospho-MBS staining was reduced to values approximately one-third of those observed in WT mice.
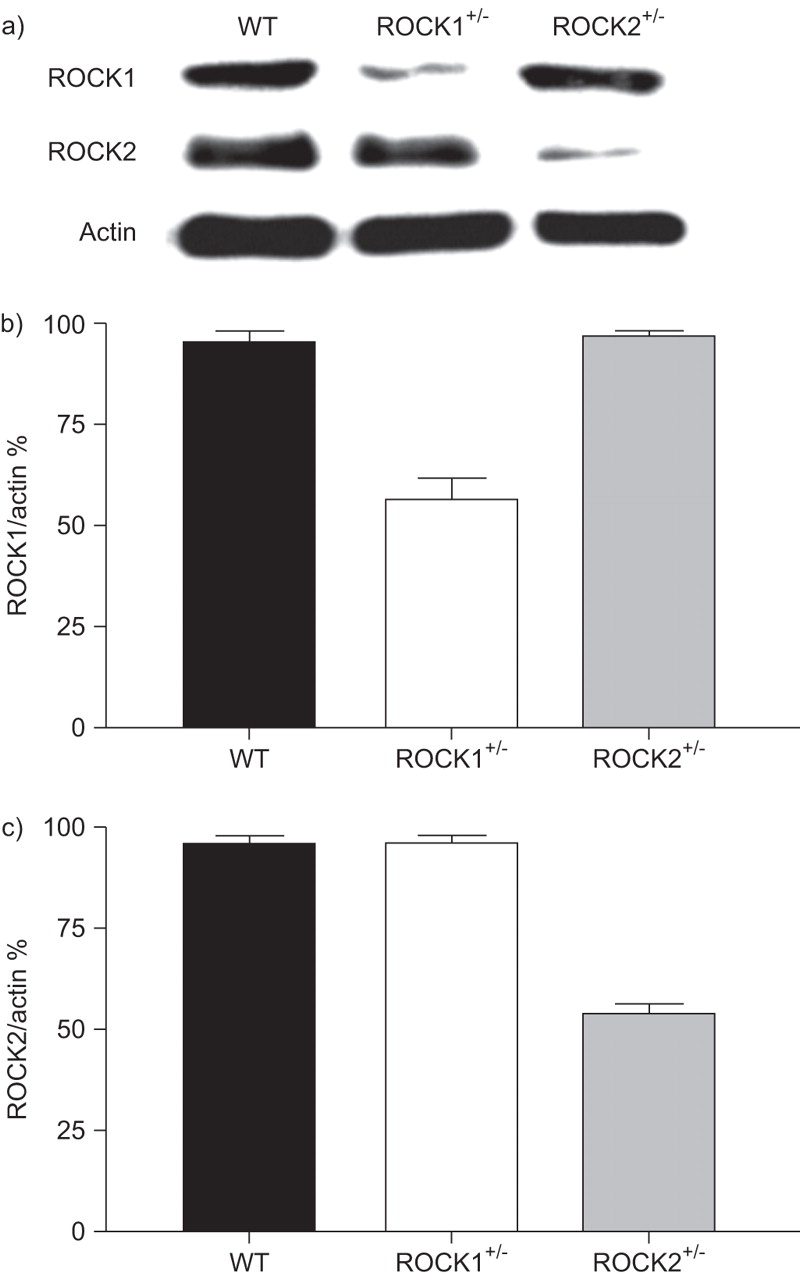
Pulmonary expression of Rho-associated coiled-coil forming kinase (ROCK) 1 and ROCK2. a) Representative western blot of ROCK1, ROCK2 and β-actin expression in lungs of wildtype (WT), ROCK1+/- and ROCK2+/- mice. b and c) Densitometric analysis showing mean±se of data from three mice in each group. ROCK1 and ROCK2 expression are normalised for β-actin expression.

Effect of Rho-associated coiled-coil forming kinase (ROCK) deficiency on ROCK activity. a) Representative western blots showing phosphorylated (phospho)-myosin-binding subunit (MBS) or total MBS in protein isolated from wildtype (WT), ROCK1+/- and ROCK2+/- mice sensitised to ovalbumin (OVA) and challenged on three consecutive days with aerosolised OVA or PBS. Lungs were harvested 48 h after the last OVA aerosol challenge. b) Densitometric analysis showing mean±se of data from three mice in each group. ROCK activity is expressed as phospho-MBS normalised for total MBS. *: p<0.05 compared with WT mice with same challenge.
Since ROCK activation had returned to near basal values by 48 h, we repeated these measurements in a separate cohort of mice in which lungs were harvested 4 h after challenge (fig. 4). Again, ROCK activation was reduced in OVA-challenged ROCK1+/- and ROCK2+/- compared with WT mice. Compared with WT mice challenged with OVA, the ratio of phospho-MBS/total MBS was reduced by 38±8% and 53±4% in ROCK1+/- and ROCK2+/- mice challenged with OVA, respectively (n=3–5 mice per group).
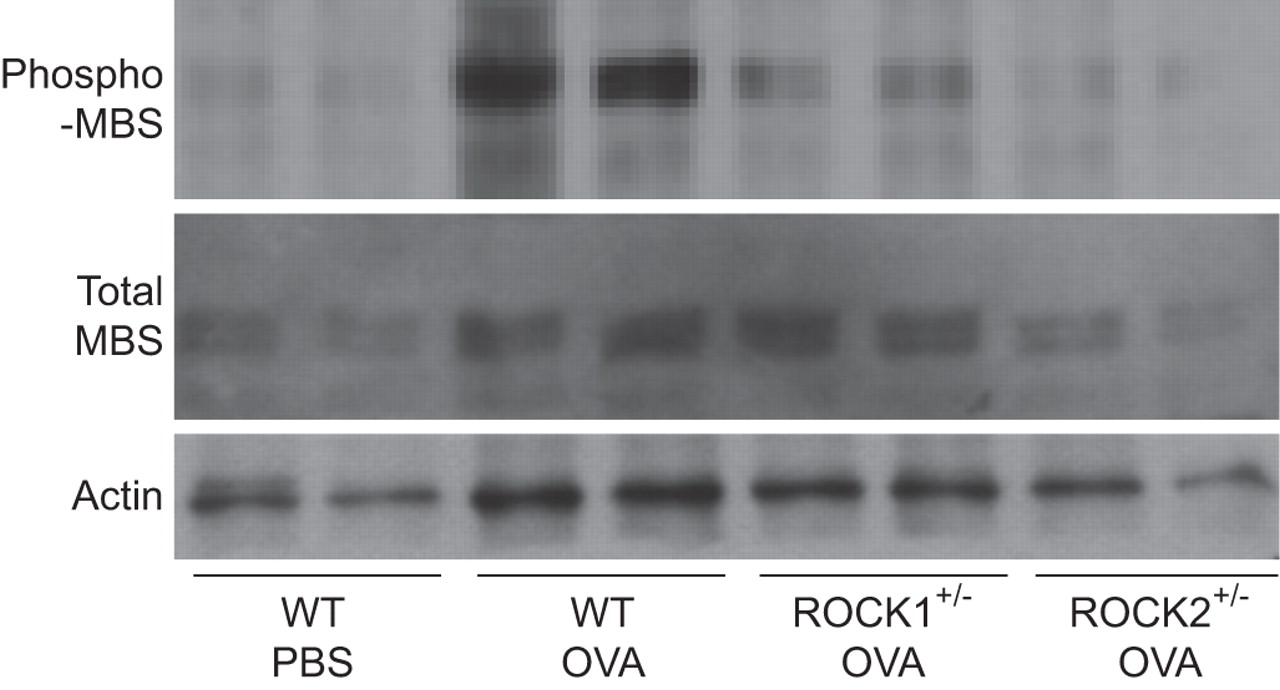
Effect of Rho-associated coiled-coil forming kinase (ROCK) deficiency on ROCK activity. Representative western blots showing phosphorylated (phospho)-myosin-binding subunit (MBS), total MBS and actin in protein isolated from wildtype (WT), ROCK1+/- and ROCK2+/- mice sensitised to ovalbumin (OVA) and challenged on three consecutive days with aerosolised OVA, along with WT mice sensitised to OVA but challenged with PBS. Lungs were harvested 4 h after the last challenge.
Pulmonary mechanics and airway responsiveness
In mice challenged with PBS, baseline RL was not significantly different in WT, ROCK1+/- and ROCK2+/- mice (table 1). Compared with PBS, OVA challenge increased RL in ROCK1+/- mice, but had no effect on baseline RL in WT or ROCK2+/- mice (table 1). Indeed, RL was significantly lower in OVA-challenged ROCK2+/- versus ROCK1+/- or WT mice (table 1).
In PBS-challenged mice, we found no differences in airway responsiveness to i.v. methacholine among WT, ROCK1+/- and ROCK2+/- mice (fig. 5a). In contrast, in otherwise untreated mice, the combined ROCK1 and ROCK2 inhibitor, fasudil, did cause a reduction in airway responsiveness to aerosolised methacholine that reached significance at 100 mg·mL−1 methacholine (fig. 5b).

Effect of Rho-associated coiled-coil forming kinase (ROCK) deficiency on baseline airway responsiveness. a) Changes in pulmonary resistance (RL) induced by i.v. methacholine in wildtype (WT), ROCK1+/- and ROCK2+/- mice sensitised to ovalbumin and challenged on three consecutive days with PBS. Results are mean±se of data from 6–8 mice in each group. b) Effect of the ROCK inhibitor, fasudil, on airway responsiveness to aerosolised methacholine. Control mice were treated with the vehicle used to dissolve fasudil, which was PBS. Results are mean of data from 6–9 mice in each group. *: p<0.05 versus PBS-treated mice.
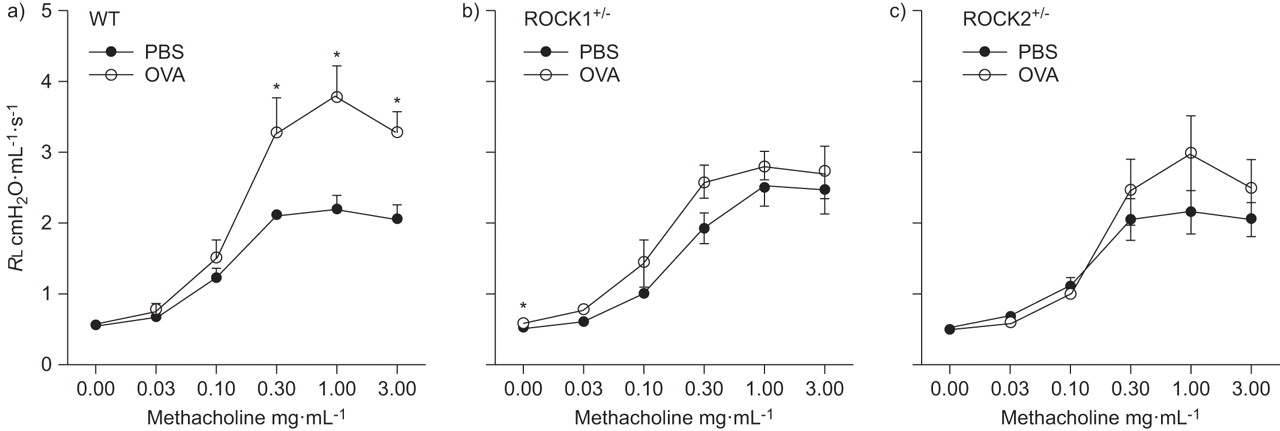
Compared with PBS, exposure to OVA caused an increase in airway responsiveness in WT mice. In contrast, OVA-induced AHR was virtually abolished in ROCK1+/- and ROCK2+/- mice (fig. 6).

Effect of Rho-associated coiled-coil forming kinase (ROCK) deficiency on ovalbumin (OVA)-induced airway responsiveness. Changes in pulmonary resistance (RL) induced by i.v. methacholine in a) wildtype (WT), b) ROCK1+/- and c) ROCK2+/- mice sensitised to OVA and challenged on three consecutive days with PBS or OVA. Mice were studied 48 h after the last challenge. Results are mean±se of data from 6–8 mice in each group. *: p<0.05 versus PBS challenge.
Pulmonary inflammation
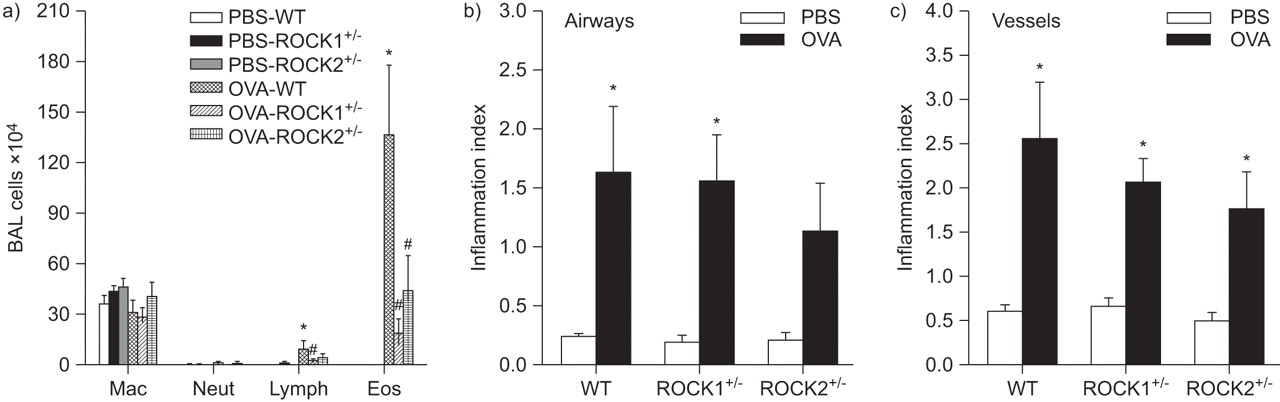
In WT mice, OVA challenge significantly increased BAL eosinophils and lymphocytes (fig. 7a). Compared with WT mice, BAL eosinophils were significantly lower in OVA-challenged ROCK1+/- and ROCK2+/- mice. A similar trend was observed for lymphocytes, but only reached statistical significance in ROCK1+/- mice. Histology also confirmed a significant increase in inflammation around both airways and vessels in OVA- versus PBS-treated mice (fig. 7b and c), but we did not observe any significant reduction in this inflammation in either ROCK1+/- or ROCK2+/- versus WT mice.

Effect of Rho-associated coiled-coil forming kinase (ROCK) deficiency on ovalbumin (OVA)-induced inflammation. a) Bronchoalveolar lavage (BAL) macrophages (Mac), neutrophils (Neut), lymphocytes (Lymph) and eosinophils (Eos) in wildtype (WT), ROCK1+/- and ROCK2+/- mice sensitised to OVA and challenged on three consecutive days with aerosolised PBS or OVA. Mice were studied 48 h after the last challenge. b) Airway and c) vessel inflammation index (see text for definition) in lungs from a separate cohort of mice sensitised and challenged in the same manner. Results are mean±se of data from 4–8 mice in each group. *: p<0.05 versus PBS-treated mice of same genotype; #: p<0.05 versus WT mice exposed to OVA.
In WT mice, BAL IL-13 and IL-5 levels increased following OVA versus PBS challenge (fig. 8). BAL IL-13 and IL-5 were not different in OVA-challenged WT and ROCK2+/- mice, although there was substantial variability in the response. In contrast, IL-13 and IL-5 were significantly reduced in OVA-challenged ROCK1+/- versus WT mice.

Bronchoalveolar lavage a) interleukin (IL)-13 and b) IL-5 (measured by ELISA) in wildtype (WT), Rho-associated coiled-coil forming kinase (ROCK)1+/- and ROCK2+/- mice sensitised to ovalbumin (OVA) and challenged on three consecutive days with aerosolised PBS or OVA. Individual values for each mouse are shown. *: p<0.05 versus PBS-treated mice of same genotype. #: p<0.05 versus WT mice exposed to OVA.
Mucous cell hyperplasia
Quantification of goblet cell number revealed that the OVA exposure protocol we employed induced robust goblet cell hyperplasia in each mouse strain when compared with strain-matched PBS-exposed control animals (fig. 9). However, after OVA treatment, both ROCK1+/- and ROCK2+/- mice had at least 40% fewer goblet cells than WT controls. Furthermore, we detected 22% fewer goblet cells in ROCK2+/- mice compared with ROCK1+/- mice (p<0.01). Goblet cell numbers in PBS-challenged mice (white bars in fig. 9g) were not affected by ROCK1 or ROCK2 insufficiency.
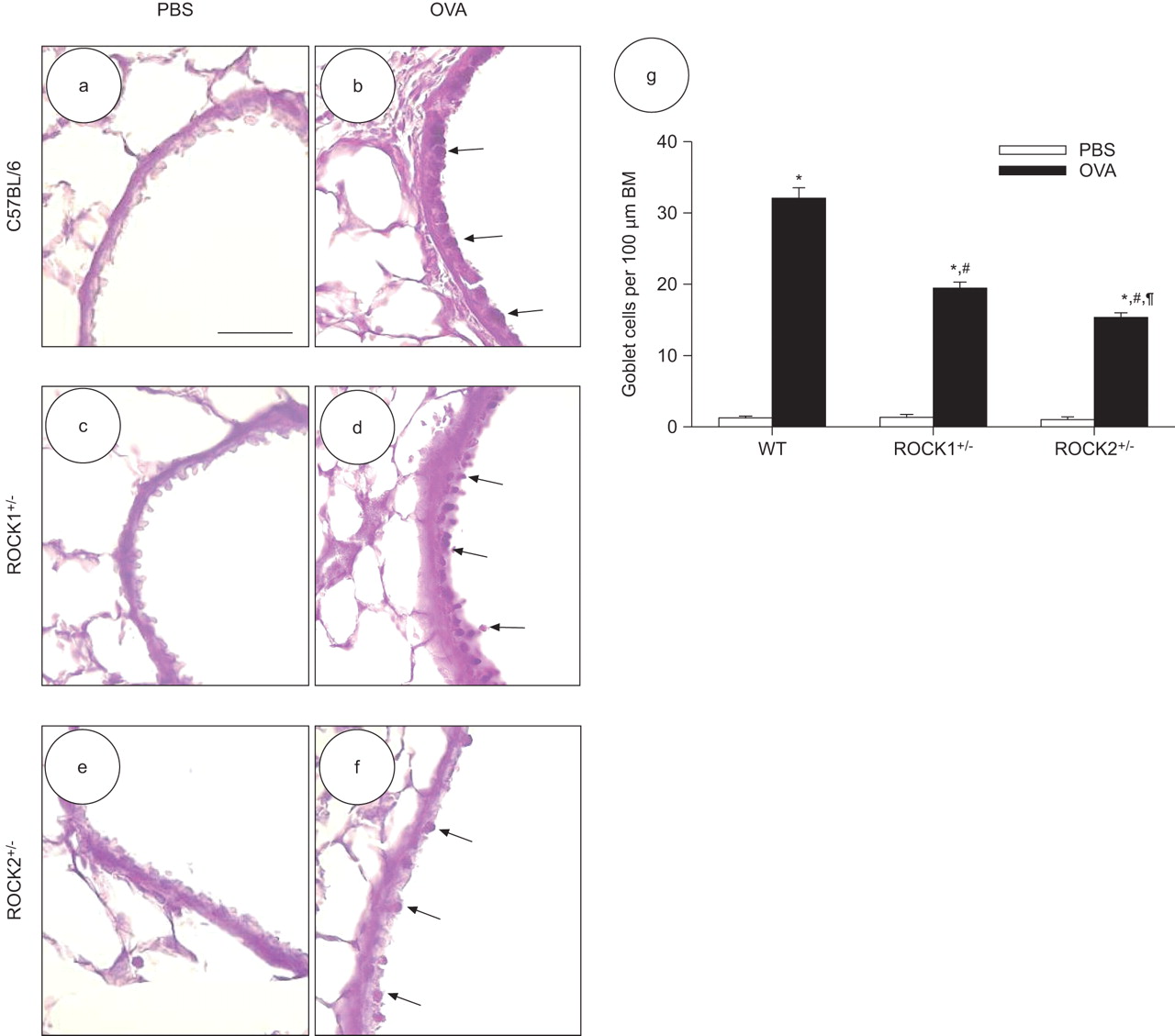
Ovalbumin (OVA)-induced goblet cell hyperplasia is reduced in Rho-associated coiled-coil forming kinase (ROCK)1+/- and ROCK2+/- mice. Representative light micrographs showing periodic acid–Schiff staining for mucus-containing goblet cells in the epithelium of medium-sized peripheral airways in mice challenged with PBS (a, c and e) or OVA (b, d and f). Representative images for wildtype (WT) C56BL/6 mice (a and b), ROCK1+/- mice (c and d), and ROCK2+/- mice (e and f) are displayed. Scale bar: 10 μm. Arrows highlight goblet cells showing positive mucus staining. g) Histogram comparing goblet cell number per 100 μm of basement membrane (BM) in peripheral airways. Data are mean±se of nine airways sampled from three different mice in each group. *: p<0.05 versus PBS-challenged mice of the same strain; #: p<0.05 compared with WT mice with the same exposure; ¶: p<0.05 compared with ROCK1+/- mice with the same exposure.
T-cell proliferation
In co-cultures of dendritic cells with T-lymphocytes from DO.11 mice that are transgenic for a T-cell receptor that recognises OVA peptide, addition of OVA caused a robust increase in lymphocyte proliferation and the ROCK inhibitor, Y-27632, caused a dose-related decrease in this proliferation (fig. 10).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Effect of the Rho-associated coiled-coil forming kinase (ROCK) inhibitor, Y-27632, on T-cell proliferation induced by culture with ovalbumin (OVA)-stimulated dendritic cells. T-cells isolated from spleens of OVA-T-cell receptor transgenic mice were incubated with dendritic cells from wildtype mice and stimulated with OVA. Tritiated thymidine was added to assess cell proliferation. Control (0 μM Y-27632) wells were treated with vehicle (cell growth medium). Results are mean±se of data from three wells in each condition. *: p<0.05 versus cells stimulated with OVA in the absence of Y-27632.
DISCUSSION
Our results indicate that inhalation of allergen in sensitised mice causes a robust increase in ROCK activation in the lungs (fig. 1). Moreover, ROCK activation appears to be functionally important, since mice with either ROCK1 or ROCK2 insufficiency had reduced allergen-induced AHR as well as decreased BAL eosinophils and fewer goblet cells following allergen challenge. Compared with WT mice, BAL lymphocytes, IL-13 and IL-5 were also significantly reduced in ROCK1+/- but not in ROCK2+/- mice. Since these ROCK1+/- and ROCK2+/- mice were only haploinsufficient in a single ROCK isoform, the results also indicate independent requirements for both ROCK1 and ROCK2 in allergic airways responses.
To our knowledge, this is the first definitive demonstration of ROCK activation in the lungs following allergen challenge. To assess ROCK activation we used western blotting of lung homogenates with an antibody that recognises phosphorylation of MBS at Thr853. With respect to specificity, ROCK has been shown to specifically phosphorylate Thr853 [23], while other kinases are capable of phosphorylating multiple other amino acid positions of MBS. Indeed, we have previously reported that western blotting using this phospho-Thr853 MBS antibody correlates closely with ROCK activity as determined by a radiolabelled kinase assay [24]. Our results show that there was some constitutive activation of ROCK in the lungs of PBS-challenged mice (figs 1, 3] and 4]; PBS groups). However, inhalation of OVA caused a rapid increase in ROCK activity in the lungs: increased MBS phosphorylation (compared with PBS inhalation) was observed as soon as the mice were removed from the OVA exposure chamber (time 0 in fig. 1). Over the next few hours, ROCK activity continued to increase, peaking at about 2–4 h and then declining.
RhoGEFs are activated by many of the mediators of allergic asthma including certain cytokines, leukotrienes, endothelin, lysophosphatidic acid and sphingosine-1-phosphate (S1P) [3]. Activation of RhoGEFs leads to GTP binding to RhoA, a process that leads to ROCK activation. RhoA protein expression has been shown to be increased in bronchi of allergen-sensitised and -challenged rats and guinea pigs [4, 5], perhaps via induction of IL-13, since IL-13 administration either in vivo or in vitro also increases RhoA expression in mouse airways [32]. It is likely that this increased RhoA expression amplifies the effects of RhoGEF activation that result in ROCK activation following OVA challenge.
To examine the functional significance of OVA-induced ROCK activation, we used heterozygous ROCK1 and ROCK2 deficient mice. Our results indicate a ∼50% reduction in pulmonary expression of ROCK1 in ROCK1+/- mice with no compensatory change in ROCK2 (fig. 2). Similarly, ROCK2+/- mice exhibited a ∼50% decrease in pulmonary ROCK2 expression with no effect on ROCK1. Similar reductions in ROCK1 or ROCK2 have been demonstrated in other tissues in these mice [20, 21]. Taken together, the data indicate that compensatory changes in expression of one ROCK isoform do not occur in response to loss of the other isoform. Thus, use of these mice allows us to separately evaluate the distinct role of each ROCK isoform in allergic airways responses. This approach offers considerable advantage over using pharmacological inhibitors that do not exhibit ROCK isoform specificity. Importantly, in our mice, declines in ROCK1 or ROCK2 expression also resulted in decreased ROCK activity (figs 3 and 4]).
One of the best described roles for ROCK is in contraction of smooth muscle [33]. Contraction of smooth muscle requires the phosphorylation of the myosin regulatory light chain (MLC). As described above, ROCK phosphorylates one of the subunits of MLCP, inhibiting its activity. Thus, for a given change in intracellular Ca2+ caused by a contractile agonist, ROCK augments MLC phosphorylation and augments contraction induced by a variety of spasmogens including cholinergic agonists, endothelin, S1P, histamine and prostaglandin F2α [4, 32–35]. Because of the role of ROCK in smooth muscle contraction, we examined its effects on baseline airway responsiveness. In PBS-challenged mice, we found no differences in airway responsiveness among WT, ROCK1+/- and ROCK2+/- mice (fig. 5a). However, we did observe a significant reduction in responsiveness when we examined the effect of a combined ROCK1 and ROCK2 inhibitor, fasudil, in unchallenged mice (fig. 5b), consistent with reports of others using the ROCK inhibitor Y-27632 [6, 36]. Taken together, the data suggest that, in the unchallenged state, one ROCK isoform can compensate for reductions in the other, and only when both are disrupted (e.g. with fasudil) does one see effects on airway responsiveness. However, this is not the case in the allergic airway. Compared with PBS, exposure to OVA caused an increase in airway responsiveness in WT mice. This OVA-induced AHR was virtually abolished in both ROCK1+/- and ROCK2+/- mice (fig. 6). These results are consistent with previous reports showing that pharmacological inhibitors of ROCK attenuate airway responsiveness to methacholine or serotonin in OVA-sensitised and -challenged mice [6, 9, 36]. Our results extend these observations by demonstrating that both ROCK1 and ROCK2 are independently required for allergen-induced AHR. The data are consistent with data from the heart, indicating that only in the challenged state are the individual effects of ROCK isoforms unmasked [14].
We also observed a marked and significant reduction in BAL eosinophils after OVA challenge in both ROCK1+/- and ROCK2+/- mice (fig. 7a). ROCK can affect motility of nonmuscle cells, including eosinophils [37], since it phosphorylates not only MBS but also several other proteins involved in actin filament assembly into stress fibres [38–41]. However, as neither ROCK1 nor ROCK2 insufficiency affected the accumulation of inflammatory cells (mostly eosinophils) around airways and vessels (fig. 7b and c), eosinophil motility per se was probably not affected. In contrast to our data (fig. 7b and c), Taki et al. [9] reported a significant reduction in OVA-induced inflammatory cell infiltration around airways in mice treated with the ROCK inhibitor, fasudil. However, the effect was small: inflammatory cell infiltration was reduced by only about one-third in fasudil-treated mice, whereas, in the same study, the effect of fasudil on BAL eosinophils was quite marked. Taken together, our BAL and histological data suggest that ROCK1 and ROCK2 are independently required for transit of eosinophils across the epithelial barrier into the airspaces, perhaps as a result of ROCK activation within epithelial cells and subsequent effects on their expression of eosinophil adhesion molecules. In contrast, ROCK may be less important for eosinophil transit across the endothelium from the blood to the tissue, and for this process, effects of ROCK1 and ROCK2 may be redundant, so that only when both are inhibited (as with pharmacological inhibitors) is an effect observed.
There were fewer lymphocytes (fig. 7) and significantly lower levels of the Th2 cytokines IL-13 and IL-5 (fig. 8) in BAL fluid of ROCK1+/- versus WT mice. The reduction in Th2 cytokines has been reported by other investigators using ROCK inhibitors [9]. IL-13 is required for the AHR induced by allergen sensitisation and challenge in mice [42]. Thus, a reduction in lymphocytes producing this cytokine is sufficient to explain the lack of AHR in ROCK1+/- mice. A reduced number of lymphocytes producing Th2 cytokines is also sufficient to explain the reduction in eosinophils in the BAL fluid of ROCK1+/- mice, since IL-13 is also required for this eosinophil influx [43, 44]. It is possible that reduced ROCK1 activation in lymphocytes themselves accounts for the reduced OVA-induced lymphocyte migration observed in ROCK1+/- mice: ROCK1 activation in lymphocytes occurs in response to agents that induce lymphocyte migration, and inhibiting ROCK prevents lymphocyte chemotaxis [45]. Such results are also consistent with the observation that mice deficient in Arhgef1, a haematopoietically restricted protein with RhoGEF activity, have reduced airway inflammation and AHR following OVA sensitisation and challenge [46]. Finally, our data showing that the ROCK inhibitor Y-27632 attenuated OVA-induced lymphocyte proliferation (fig. 10) suggests that ROCK is required for the process of dendritic cell activation of T-cells. There was a trend toward inhibition of proliferation at 3 μM Y-27632 and significant and substantial inhibition at 30 μM (fig. 9). A concentration of 10 μM Y-27632 is typically effective against ROCK-dependent processes [11], so it is likely that the effects of Y-27632 that we observed (fig. 10) were ROCK dependent, but Y-27632 does inhibit other kinases at slightly higher concentrations (100 μM) [11]. Since there is evidence that dendritic cell migration can be affected by Rho kinase [47], we do not know whether Y-27632 was acting in the dendritic cells or in the lymphocytes. However, the observation that, when ROCK was inactivated, lymphocytes proliferated more slowly (fig. 10), is consistent with the reduced numbers of lymphocytes present in the BAL of ROCK1+/- mice (fig. 7).
The precise role of ROCK2 in OVA-induced AHR is less clear. Taken as a group, lymphocytes, IL-13 and IL-5 were not reduced in BAL of ROCK2+/- mice (figs 7a and 8), although there was substantial variability in the response: several mice had little or no Th2 cytokines, whereas others had BAL Th2 cytokine levels similar to WT mice. The results suggest that there may be different roles for ROCK2 versus ROCK1 in this model. Consistent with this hypothesis, others have reported differences in both the regulation and localisation of ROCK1 and ROCK2 [48]. Eosinophils were reduced in the BAL of ROCK2+/- mice, and it is conceivable that this reduction in eosinophils is responsible for the lack of AHR observed in ROCK2+/- mice, although the role of eosinophils in allergen-induced AHR remains controversial, even in mice [43, 49]. Moreover, eosinophils were not reduced in the airway tissue. Alternatively, there were substantial reductions in baseline RL in OVA-challenged ROCK2+/- versus WT or ROCK1+/- mice (table 1), suggesting that effects of ROCK2 on airway smooth muscle may contribute to its role in OVA-induced AHR.
OVA challenge caused goblet cell hyperplasia (fig. 9). Importantly, goblet cell hyperplasia was greater in WT than in either ROCK1+/- or ROCK2+/- mice (fig. 9), indicating roles for both ROCK isoforms in this hyperplasia. ROCK2 deficiency had a greater effect in reducing goblet cell hyperplasia than ROCK1, suggesting a more important role for this ROCK isoform. Others have reported either a much smaller (∼15% reduction) or no effect of the ROCK inhibitors fasudil or Y-27632 on OVA-induced mucous cell hyperplasia in BALB/c mice [6, 9]. The greater importance of ROCK in this study may be related to mouse strain, since we used C57BL/6 mice. Alternatively, it is conceivable that there are nonspecific effects of these ROCK inhibitors that counter the effects of ROCK inhibition.
In summary, we report ROCK activation in the lungs following OVA challenge in OVA-sensitised mice. Using isoform-specific heterozygous mice, our data demonstrate distinct requirements for both ROCK1 and ROCK2 in allergic airway responses. Understanding the precise role of ROCK1 and ROCK2 in animal models of asthma could lead to new strategies for intervention in this highly prevalent disease.
Footnotes
Support Statement
This study was supported by National Heart, Lung, and Blood Institute Grants HL-084044, HL-052233 and HL-091933, and by National Institute of Environmental Health Sciences Grant ES-013307. P-Y. Liu was supported by the National Health Research Institute of Taiwan. Work completed by A.J. Halayko was supported by Canadian Institutes of Health Research Grant FRN 77759, the Canada Research Chairs Program, and by the Manitoba Institute of Child Health.
Statement of Interest
Statements of interest for A.J. Halayko, S.A. Shore and J.K. Liao can be found at www.erj.ersjournals.com/site/misc/statements.xhtml
- Received August 4, 2010.
- Accepted April 5, 2011.
- ©ERS 2011
REFERENCES










