





Abstract
Pulmonary veno-occlusive disease (PVOD) is a rare form of pulmonary hypertension (PH) characterised by preferential remodelling of the pulmonary venules. In the current PH classification, PVOD and pulmonary capillary haemangiomatosis (PCH) are considered to be a common entity and represent varied expressions of the same disease. The recent discovery of biallelic mutations in the EIF2AK4 gene as the cause of heritable PVOD/PCH represents a major milestone in our understanding of the molecular pathogenesis of PVOD. Although PVOD and pulmonary arterial hypertension (PAH) share a similar clinical presentation, with features of severe precapillary PH, it is important to differentiate these two conditions as PVOD carries a worse prognosis and life-threatening pulmonary oedema may occur following the initiation of PAH therapy. An accurate diagnosis of PVOD based on noninvasive investigations is possible utilising oxygen parameters, low diffusing capacity for carbon monoxide and characteristic signs on high-resolution computed tomography of the chest. No evidence-based medical therapy exists for PVOD at present and lung transplantation remains the preferred definitive therapy for eligible patients.
Abstract
Recent advances such as discovery of the genetic basis of PVOD will pave way for future translational research http://ow.ly/YldhC
Introduction
Pulmonary hypertension (PH) is a haemodynamic state of the pulmonary circulation defined by a mean pulmonary artery pressure (mPAP) ≥25 mmHg [1]. Pulmonary veno-occlusive disease (PVOD) represents a rare form of PH characterised by preferential involvement of the pulmonary venous system [2, 3]. The pathological hallmark is obliteration of small pulmonary veins by fibrous intimal thickening and patchy capillary proliferation [4]. Similar to other types of obstructive pulmonary vascular diseases, PVOD results in a progressive increase in pulmonary vascular resistance, culminating in right heart failure and death [5].
The first detailed clinical and pathological description of PVOD was attributed to the German physician J. Hora more than 80 years ago [6]. However, it was not until 1966 that the term of PVOD was coined by Heath et al. [7], who recognised that PVOD was a distinct entity from so-called “primary pulmonary hypertension” (now termed idiopathic pulmonary arterial hypertension (PAH)). Recently, a major milestone in our understanding of PVOD was achieved with the discovery that biallelic mutations of the EIF2AK4 (eukaryotic translation initiation factor 2 alpha kinase 4) gene are responsible for heritable PVOD [8]. Despite significant strides in our knowledge of the genetic, cellular and molecular basis of PVOD over the past decade, it remains classically an orphan lung disease without any proven efficacious drug therapies. Lung transplantation remains the destination therapy for eligible patients. This review aims to provide a comprehensive state-of-the art update on PVOD.
PVOD in the current PH classification system
The current classification system from the fifth World Symposium on PH (Nice, 2013) and the 2015 European Society of Cardiology (ESC)/European Respiratory Society (ERS) guidelines on the diagnosis and management of PH divides PH into five main groups according to shared pathophysiology, clinical features and therapeutic approaches (table 1) [1, 5, 9]. Group 1 PH consists of PAH and its various subtypes such as idiopathic, heritable and drug-induced PAH as well as those associated with connective tissue disease, HIV infection, portal hypertension, congenital heart disease and schistosomiasis. Although PVOD is classified as belonging to group 1 PH, it has been given a special subgroup designation of group 1′ under the unified entity of PVOD/pulmonary capillary haemangiomatosis (PCH). Previously regarded by some as different entities, current evidence supports the concept that PVOD and PCH are in fact varied expressions of the same disorder. Indeed, clinico-pathological studies indicate marked overlap in the histological findings of PVOD and PCH [10], and their clinical and radiographic findings are virtually indistinguishable [11–13]. The notion of a common disorder is further emphasised by the recent discovery that mutations in the EIF2AK4 gene are responsible for heritable cases of both PVOD and PCH [8, 14].
Classification of pulmonary hypertension according to European Society of Cardiology/European Respiratory Society Guidelines
PVOD and PCH have been given the subgroup designation of group 1′ in order to highlight both similarities and important differences with PAH. In general, the clinical presentation and haemodynamic findings of PVOD/PCH and PAH are similar, but the clinical course of PVOD/PCH is usually more aggressive [15]. Importantly, PVOD/PCH typically demonstrate a poor response to PAH therapy and the use of PAH drugs is associated with a potential risk of life-threatening pulmonary oedema [16]. The 2015 ESC/ERS guidelines [1] have proposed further refinements to the classification of group 1′ PVOD/PCH with an expanded subclassification to include idiopathic, heritable, drug and toxins-induced, and associated forms of PVOD (table 1). As PVOD and PCH are currently considered to be the same disorder, we have chosen to use the term PVOD to represent the entity of PVOD/PCH throughout this review.
Histological features
The defining pathological feature of PVOD is the diffuse involvement of venules and septal veins in a vasculopathy characterised by intimal fibrosis resulting in luminal narrowing or obliteration (figure 1) [4, 17]. The massive involvement of small pre-septal venules with occlusive or near occlusive fibrous thickening of the intima is usually considered necessary for the diagnosis of PVOD, since moderate fibrous changes and “arterialisation” of pulmonary veins can be seen in other forms of long standing pulmonary venous hypertension such as mitral stenosis [17]. In PVOD, intimal remodelling of veins and venules may range from loose fibrous tissue with variable cellularity to dense, paucicellular, sclerotic lesions. It is conceivable that such variations reflect different stages in the evolution of vascular lesions. Elastic fibres within the venous wall may display calcium incrustations, which appear as black pigment-like deposits. Small granulomas containing giant cells that show phagocytosis of degraded elastic fibres are regularly encountered in the vicinity of small veins and sometimes arteries.

Vascular lesions and pathological features of pulmonary veno-occlusive disease (PVOD). All three compartments of the pulmonary microcirculation are affected in PVOD, although there is a preferential involvement of the pulmonary venous system. Venular lesions include intimal fibrosis of small pre-septal venules. Capillary lesions are characterised by exuberant proliferation of endothelial cells (capillary haemangiomatosis). Arterial lesions resemble those of pulmonary arterial hypertension with intimal fibrosis and medial hypertrophy, but complex plexiform lesions are absent.
Alveolar capillaries in PVOD are typically dilated and engorged due to downstream obstruction, and frank angioproliferation may even occur to produce capillary lesions indistinguishable from PCH (figure 1) [18]. Pulmonary and pleural lymphatics are also dilated. In a pathological series of specimens from 35 patients diagnosed with either PVOD (n=30) or PCH (n=5), Lantuejoul et al. [10] demonstrated that 73% of PVOD patients displayed PCH-like angioproliferative lesions. Occluding venous lesions were also found in the majority of patients diagnosed with PCH. These findings suggest that, in most cases, PCH represents a secondary angioproliferative process caused by post-capillary obstruction.
Although post-capillary and capillary remodelling is usually more conspicuous, pulmonary arterial lesions are also encountered in PVOD. Arterial lesions in PVOD resemble that of PAH with eccentric intimal fibrosis and medial hypertrophy (figure 1). However, plexiform (or complex) lesions characteristic of severe PAH are not present. Thrombotic occlusion can complicate vascular lesions and subsequent recanalisation may produce “colander-like” lesions. Occult pulmonary haemorrhage occurs frequently in PVOD, probably due to post-capillary obstruction [19]. Large amounts of haemosiderin are found in alveolar macrophages and type 2 pneumocytes as well as deposits in the interstitium. Oedema is also present in the interstitium and is most prominent along the interlobular septae, which may eventually progress to fibrosis.
In an explanatory approach, different forms of chronic pulmonary vascular diseases such as PAH, PVOD or chronic thromboembolic PH have traditionally been labelled by their anatomical site of predilection, e.g. muscular pulmonary arteries, pulmonary venules or larger elastic type arteries. However, recent pathological studies have led to the increasing recognition of an apparent overlap between arterial and venous diseases [20]. Dorfmüller et al. [21] showed that 75% of patients with connective tissue disease-associated PAH display significant obstructive venous lesions. This finding may explain, in part, the clinical observation of poor response to PAH therapy in some patients with systemic sclerosis-associated PAH. Intriguingly, a recent study by the same authors has shown that conspicuous remodelling of pre-septal venules, septal veins and systemic lung vessels are also present in patients with chronic thromboembolic PH, suggesting that post-capillary remodelling may be related to the increase in patent bronchopulmonary venous anastomoses [22].
Genetics of PVOD
In 2014, the French PH network reported the genealogy of 13 PVOD families with heritable disease [8]. With careful inspection of the genealogy of these 13 families, we were struck by the fact that only one generation was affected by the disease and a high frequency of consanguinity was encountered, which was present in more than half of the families (figure 2). Both of these observations are characteristic of autosomal recessive transmission. Whole-exome sequencing on two affected siblings from five families allowed the demonstration that biallelic mutations in EIF2AK4 were present in all 13 PVOD families. Interestingly, one of the 13 families carrying EIF2AK4 mutations was initially diagnosed with PCH [8]. Subsequently, Best et al. [14] also identified EIF2AK4 mutations in another PCH family and in sporadic PCH cases. More recently, large gypsy families from Spain have been described with several cases of PVOD due to bi-allelic EIF2AK4 mutation [23]. Paediatric cases of PVOD associated with biallelic mutations of EIF2AK4 have been also reported and, to date, the youngest patient diagnosed with heritable PVOD was 11 years old [8, 14, 23].
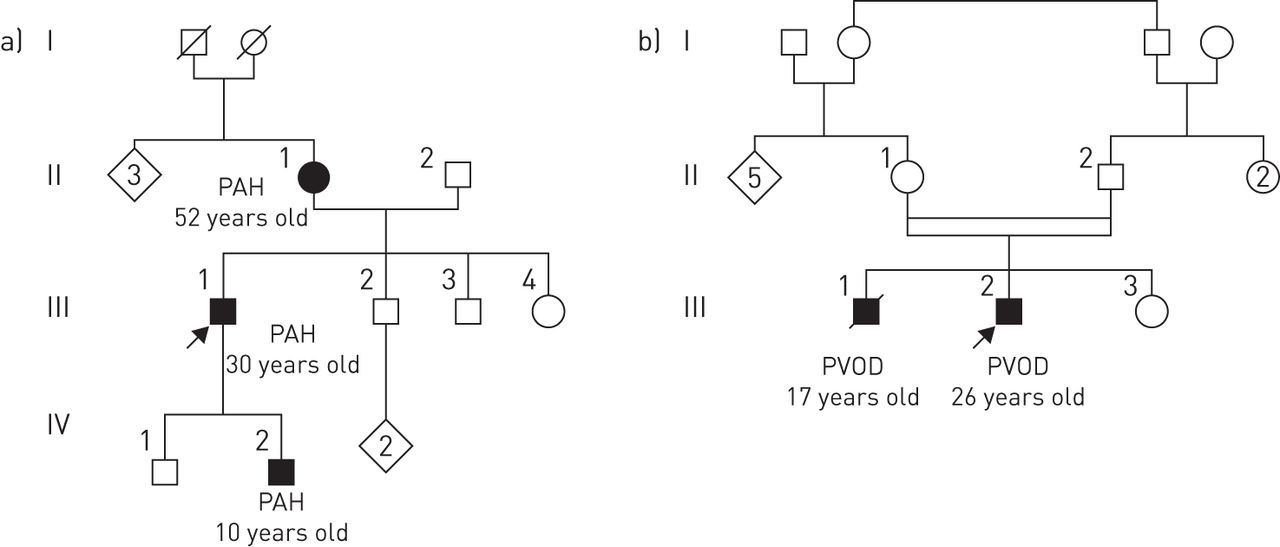
Illustrative family trees of heritable pulmonary arterial hypertension (PAH) and pulmonary veno-occlusive disease (PVOD). a) Heritable PAH due to a BMPR2 (bone morphogenetic protein receptor type 2) mutation and b) heritable PVOD due to biallelic mutations in the EIF2AK4 (eukaryotic translation initiation factor 2 alpha kinase 4) gene. Heritable PAH is transmitted as an autosomal dominant trait with incomplete penetrance; each generation could be affected by the disease. Heritable PVOD is transmitted as an autosomal recessive trait; only one generation is affected by the disease and consanguinity is often reported.
In the French PH network, genetic counselling and EIF2AK4 mutation screening have been offered to all patients with confirmed or suspected PVOD, with or without a family history of the disease [24]. Although biallelic EIF2AK4 mutations had been found in all familial cases of PVOD, mutations were also identified in 9% (7 out of 81) of apparently sporadic cases of PVOD. EIF2AK4 mutation carriers are characterised by a young age at diagnosis and the absence of risk factors for PVOD, such as exposure to alkylating agents or organic solvents (see later in this review). To date, the penetrance of PVOD in carriers of biallelic EIF2AK4 mutations appears high, but the exact rate of penetrance remains to be determined.
Finally, the discovery of EIF2AK4 mutation confirmed that PVOD and PCH are two clinico-pathological presentations of the same disease entity. All patients carrying biallelic mutations in the EIF2AK4 gene are grouped under the term “heritable”, group 1′.2 in the current classification of PH (table 1). Although mutations in the bone morphogenetic receptor type 2 (BMPR2) gene have been reported in familial forms of suspected PVOD [25], these cases of PVOD associated with BMPR2 mutations raise the question of possible misclassification of these patients.
Risk factors and associated conditions in PVOD
Chemotherapy
Numerous case reports have reported a temporal association between administration of chemotherapeutic agents and the development of PVOD [26–35]. Hepatic veno-occlusive disease (or sinusoidal obstruction syndrome) is an uncommon but well-recognised complication of high-dose chemotherapy, particularly in the setting of haematopoietic stem cell transplantation [36, 37]. Ranchoux et al. [38] recently performed a systematic analysis of possible cases of chemotherapy-induced PVOD from the French PH network and concluded that alkylating agents represented the predominant drug class associated with chemotherapy-induced PVOD. Unfortunately, most of the patients received a regimen of chemotherapy that included several concomitant or sequential drugs, resulting in difficulties in identifying specific drugs that were associated with PVOD. The French PH network have also recently reported on cases of PVOD in patients receiving mitomycin-C alone or in association with 5-fluorouracil for the treatment of squamous anal cancer [39]. Using data from cancer registries in France, the estimated incidence of PVOD in patients with anal cancer was 3.9 per 1000 patients per year, markedly higher than the incidence rate of 0.5 per million persons per year for idiopathic PVOD [39]. Chemotherapy-induced PVOD has been reported in the setting of paediatric cancer and haematological malignancies, particularly after bone marrow transplantation [30, 40]. Radiotherapy has been suggested as a risk factor for PVOD, but data are lacking. Of note, the identification of alkylating agents as a risk factor for PVOD has allowed the development of novel animal models of PVOD (see later).
Organic solvent exposure
Occupational exposure is emerging to be a potentially relevant and frequently encountered risk factor for PVOD. In a recent case–control study comparing PVOD patients with PAH [41], PVOD was linked to occupational exposure to organic solvents, with trichloroethylene (a chlorinated solvent) being the main agent implicated in this association. A history of significant trichloroethylene exposure was found in 42% of PVOD patients versus only 3% of PAH (adjusted OR 8.2, 95% CI 1.4–49.4). PVOD patients with a positive exposure history were characterised by older age of disease onset and the absence of mutations for the EIF2AK4 gene, whereas those without an exposure history were typically younger and a significant proportion harboured biallelic EIF2AK4 mutations [41].
Tobacco exposure
Cumulative tobacco exposure has been reported to be higher in PVOD compared with idiopathic PAH, a finding that is independent of gender [25]. In the recent study demonstrating the role of organic solvent in the development of PVOD, it is important to note that all patients with significant exposure to trichloroethylene had concurrent tobacco exposure (mean total exposure of 38 pack-years) [41]. This could suggest a potentiator effect of tobacco and solvent exposures for the development of PVOD. Interestingly, a higher frequency of tobacco exposure has also been found in patients with PAH compared with healthy controls or chronic thromboembolic PH patients [42]. It is well known that tobacco exposure can result in endothelial dysfunction [43] and can directly induce pulmonary vascular remodelling in animal models of PH [44, 45]. However, it remains undetermined why tobacco exposure appears to be more strongly associated with PVOD, which has a predilection for affecting the pulmonary venous and capillary compartments.
Autoimmunity and inflammatory conditions
It is increasingly recognised that significant venular involvement may be a relatively common occurrence in connective tissue disease-associated PAH, particularly systemic sclerosis [21, 46, 47]. PVOD has also been reported to be associated with other inflammatory disorders such as sarcoidosis [48], Langerhans' cell granulomatosis [49] and Hashimoto's thyroiditis [50], although these associations are restricted to isolated case reports or small series. The true prevalence of PVOD in connective tissue disease-associated PAH is unknown, as sufficiently large histological series are unavailable for systematic evaluation. However, a recent study suggested that as many as two-thirds of patients with precapillary PH due to systemic sclerosis may display radiological signs of PVOD on high-resolution computed tomography (HRCT) of the chest [51], consistent with the estimation of proportion of significant venous involvement in histological series of connective tissue disease-associated PAH [21].
Molecular pathogenesis and animal models of PVOD
In contrast to PAH, little is currently known regarding the pathobiology of PVOD. However, PVOD and PAH display some overlapping clinical and histological features, and it is likely that they share certain convergent molecular pathways. Indeed, both conditions are characterised by over-activation of the platelet-derived growth factor/platelet-derived growth factor receptor axis [52–55] and serotonin-induced smooth muscle hyperplasia is a common pathogenic mechanism seen in various forms of PH [56]. In both PVOD and PAH, endothelial proliferation is a feature of vascular lesions and proangiogenic factors producing c-kit+ mast cells also infiltrate vascular lesions [53, 54, 57]. Although controversial, another common molecular pathomechanism between PAH and PVOD may be the decreased production of nitric oxide (NO) by the pulmonary endothelium. Indeed, endothelial NO synthase is specifically reduced in PVOD when pulmonary arterial hypertensive remodelling is concomitantly present [58]. However, in contrast to plexiform lesions of idiopathic PAH, vascular lesions of PVOD retain markers of cell growth suppression such as peroxisome proliferation-activated receptor-γ and caveolin-1. This suggests that the atypical endothelial cell phenotypes that lead to PVOD and idiopathic PAH are distinct [59].
Immune-mediated injury of pulmonary venules has been proposed as a potential mechanism and inflammatory conditions are known to be associated with PVOD. Cytotoxic T-cell, natural killer cell and natural killer T-cell functions may play a critical role in vascular homeostasis. The analysis of the cytolytic compartment of inflammatory cells showed significant modifications in the proportions of circulating cytotoxic T-cell, natural killer cell and natural killer T-cell populations in PVOD and not in PAH, and this is associated with wasting of their granulysin content in PVOD [60]. This may explain the rise in the granulysin serum concentration in PVOD as compared with PAH patients. Granulysin is implicated in a myriad of diseases including infection, cancer, autoimmunity, skin and reproductive diseases [61], and its deregulation in PVOD may also be of importance in the pathogenesis of the disease.
The discovery of biallelic mutations in the EIF2AK4 gene as the cause of heritable PVOD has created opportunities to elucidate the central molecular pathways involved in PVOD pathogenesis, akin to our understanding of the central role of the BMPR2/transforming growth factor-β axis in PAH following the discovery of BMPR2 mutations. The EIF2AK4 gene codes for general control nonderepressible 2 (GCN2), which belongs to a family of four kinases that phosphorylate the α-subunit of eukaryotic translation initiation factor 2 (eIF2α) (figure 3). These four serine/threonine kinases, GCN2, PERK (PKR-like endoplasmic reticulum kinase), PKR (protein kinase double-stranded RNA-dependent) and HRI (heme-regulated inhibitor), are involved in control of general translation in response to various cellular stresses. Phosphorylation of eIF2α protects cells by reducing protein synthesis and regulating changes in expression of genes involved in stress responses, defined as the “integrated stress response” [62, 63]. These genes are involved in a variety of cellular mechanisms, such as resistance to oxidative stress, inflammation, cell survival or angiogenesis. In particular, GCN2 is known to induce changes in gene expression in response to amino acid deprivation (figure 3) [8, 62, 63]. GCN2 has been demonstrated to be involved in responses to other triggers, including viral infections, hypoxia or ultraviolet radiation [63]. To date, eIF2α is the only characterised substrate of GCN2, but there is evidence that the effects of activation of GCN2 could occur without changes in eIF2α phosphorylation, suggesting other pathways may be controlled by GCN2. The role and expression of GCN2 in the pulmonary vasculature is largely unknown; however, one can hypothesise that a reduction of GCN2 activity due to EIF2AK4 mutations may lead to an increase in vulnerability to oxidative stress and an increase in inflammation [64]. Regarding the venular and capillary toxicity of alkylating agents, it is of interest to note that in animal models (Gcn2-/- mice), a loss of GCN2 promotes oxidative stress and inflammation-mediated DNA damage during asparaginase therapy, suggesting that GCN2 protects against hepatotoxicity during asparaginase treatment [64]. Hence, further investigations will be needed to understand the link between EIF2AK4 mutations, GCN2 expression and the development of venular and capillary lesions in human PVOD.
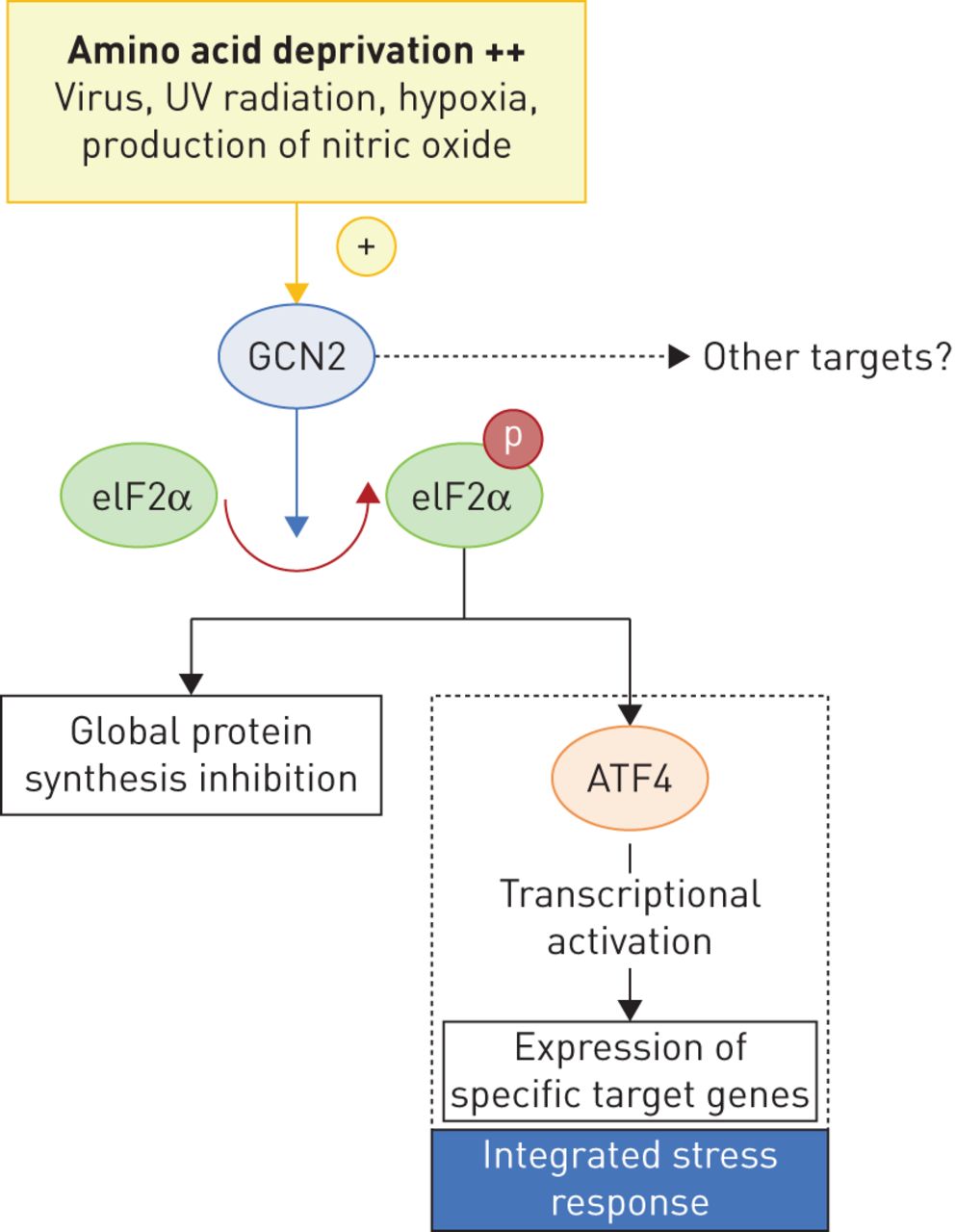
GCN2 (general control nonderepressible 2) pathway. Heritable pulmonary veno-occlusive disease (PVOD) is due to biallelic mutations of the EIF2AK4 (eukaryotic translation initiation factor 2 alpha kinase 4) gene coding for GCN2. GCN2 belongs to a family of four kinases and phosphorylates the α-subunit of eukaryotic translation initiation factor 2 (eIF2α), which is involved in control of general translation in response to various cellular stresses (via activating transcription factor (ATF)4). The main trigger for GCN2 activation is amino acid deprivation (++), but it has been demonstrated that GCN2 may be involved in response to other triggers, such as viral infections, hypoxia, production of nitric oxide (decreased arginine) or ultraviolet (UV) radiation. Phosphorylation of eIF2α protects cells by reducing protein synthesis and regulating changes in expression of genes involved in stress responses (resistance to oxidative stress, inflammation, cell survival or angiogenesis), defined as the “integrated stress response”. The link between EIF2AK4 mutations, GCN2 expression and the development of venular and capillary lesions in human PVOD remains elusive.
Recently, Lathen et al. [65] demonstrated that the ERG–apelin receptor (APLNR) axis may play a role in the control of pulmonary venule endothelial proliferation. ERG is a member of the Ets family of transcription factors and has been identified as a modulator of endothelial cell differentiation. These authors demonstrated that ERG binds to and serves as a transcriptional activator of the G-protein-coupled receptor gene, APLNR, and that knockout of either ERG or APLNR results in pulmonary venous specific endothelial proliferation in vitro. Mice with homozygous deletion of ERG or ALPNR develop remodelled post-capillary pulmonary venules. In human PVOD lung tissue, levels of ERG protein and APLNR mRNA and protein were significantly decreased compared with control lungs. However, the authors did not find any difference in the expression of GCN2 in lung tissues from animals with homozygous or heterozygous deletion of ERG or APLNR. Hence, the relationship between EIF2AK4 and the ERG–APLNR axis should be better characterised to understand the pathobiology of PVOD.
With the identification of alkylating agents as a risk factor for the development of PVOD [38], animal models of chemotherapy-induced PH have been explored. In experimental models, cyclophosphamide exposure was able to induce PH in three different animal species: mouse, rat and rabbit [38]. In rats, intraperitoneal injection of mitomycin-C resulted in the development of PVOD, as demonstrated by the presence of PH at right heart catheterisation and preferential remodelling of small pulmonary veins together with foci of intense endothelial cell proliferation of the capillary bed [39]. Importantly, mitomycin-C administration resulted in a dose-dependent depletion of lung GCN2 protein levels whereas lung BMPR2 levels were unaffected in these animals. Animal models will provide an opportunity to gain further insights into the molecular mechanism of PVOD and the exploration of novel therapeutic targets.
Epidemiology
Prevalence and incidence rates of PVOD can only be estimated since accurate diagnosis is challenging without histological (or genetic) confirmation and many cases of PVOD remain misclassified as PAH. Histopathological series of patients with hypertensive pulmonary vascular disease suggest that PVOD accounts for perhaps 3–12% of cases labelled as “idiopathic PAH” ante-mortem [66–68]. The representation of PVOD might be even higher if patients with “treatment refractory PAH” who require lung transplantation are considered [69]. Thus, a conservative estimate is a prevalence of ∼1–2 cases per million inhabitants with an annual incidence rate of ∼0.1–0.5 per million for idiopathic or heritable PVOD. However, this figure does not account for PVOD in the setting of connective tissue disease or other associated conditions/risk factors (chemotherapy, solvents), suggesting that the true number of PVOD cases is likely to be higher.
PVOD has been reported to occur across all age groups from paediatric to elderly populations [3, 19, 25]. Discovery of biallelic mutations of the EIF2AK4 gene in 13 families from the French PH network suggests that heritable PVOD patients are young at diagnosis (27±10 years) [8]. As suspected with autosomal recessive transmission, the sex ratio in heritable PVOD is equally distributed [25, 70], which is in contrast to the female predominance found in PAH [71, 72]. Sporadic PVOD cases are predominantly male with an older age at diagnosis [41].
Diagnosis of PVOD
Clinical features
PVOD and PAH share a common clinical presentation and are characterised by insidious onset of fatigue and breathlessness progressing to symptoms of right heart failure in end-stage disease. Similar to PAH, diagnosis is often delayed and most patients are in New York Heart Association functional class III or IV by the time of formal diagnosis [25, 73, 74]. Typical clinical signs of PH are found with right ventricular heave, a loud pulmonary component of the second heart sound and a systolic murmur of tricuspid regurgitation together with signs of overt right heart failure in decompensated disease. Auscultatory crackles over the lung fields and pleural effusions may be present but these features are rare apart from the situation of pulmonary oedema complicating pulmonary vasodilator therapy. The development of pulmonary oedema following vasodilator therapy strongly supports the diagnosis of PVOD. Despite the presence of alveolar haemorrhage on histological examination and bronchoalveolar lavage (BAL), clinically apparent haemoptysis is not more common than what is observed in PAH [25]. Clubbing and Raynaud's phenomenon are reported to occur in both PVOD and PAH in a comparable proportion [25].
Doppler echocardiography
Transthoracic echocardiography remains the most widely used technique for the detection of PH [75]. No specific features of PVOD are found on echocardiographic examination, and findings reflect the presence of precapillary PH with elevated tricuspid regurgitation velocity with or without right-sided chamber dysfunction depending on disease severity. Echocardiography may be useful to exclude left-sided cardiac disease as a cause of pulmonary oedema.
Invasive haemodynamic evaluation by right heart catheterisation
Pulmonary haemodynamics in PVOD are indistinguishable from other forms of precapillary PH and are characterised by an mPAP ≥25 mmHg, normal pulmonary artery wedge pressure (PAWP) ≤15 mmHg and elevated pulmonary vascular resistance. Although the main anatomical site of disease in PVOD resides in the post-capillary venules, PAWP is usually normal [7, 25, 70]. In a large series where patients with histologically proven PVOD and idiopathic PAH were compared, pulmonary haemodynamics at diagnosis were similar in the two groups [25].
When PAWP is obtained during balloon occlusion, flow is terminated distal to the occluded pulmonary artery branch, producing a static column of blood that reflects the pressure in a pulmonary vein of a similar diameter to that of the occluded artery branch. In other words, PAWP is a measure of the pressure in a relatively large calibre pulmonary vein, which is not affected by the disease (figure 4) [76]. In PVOD, PAWP is not a reflection of the true pulmonary capillary pressure, which is elevated in PVOD due to downstream obstruction of pulmonary venules. Elevation of pulmonary capillary pressure explains the occurrence of pulmonary oedema in PVOD, which may be precipitated by the administration of pulmonary vasodilators resulting in preferential dilatation of pulmonary arterioles and flooding of pulmonary capillaries. In clinical practice, direct measurement of pulmonary capillary pressure is not possible although analysis of the pulmonary artery pressure decay curve following balloon occlusion has been proposed as a theoretical method for estimating pulmonary capillary pressure, allowing partitioning of vascular resistance into arterial and capillary–venous components [77, 78]. However, limited studies have not demonstrated the clinical utility of this technique for the differential diagnosis of PVOD [79, 80].

Normal pulmonary artery wedge pressure in pulmonary veno-occlusive disease (PVOD). In PVOD, the small pulmonary veins are affected, leading to an elevation of pressure in this region (Pv), as well as to an elevation in true pulmonary capillary pressure (Pc) and pre-capillary pulmonary arterial pressure (Pa). Larger pulmonary veins are usually not affected by PVOD, and it is in fact the pressure here that is reflected by pulmonary artery wedge pressure; the static column of blood (hatched) occluded by pulmonary arterial catheter wedging or balloon inflation of a pulmonary arterial branch (balloon 1) reflects the pressure in a vein of similar diameter (balloon 2), usually of a larger size than those vessels affected by PVOD. Therefore, this measurement technique does not reflect the important elevation of pressure in the smaller diameter vessels associated with PVOD.
In idiopathic PAH, performing an acute vasodilator challenge is mandatory in order to identify a subset of patients (<10%) who will test positive and demonstrate a long-term response to calcium channel blockers [81, 82]. Furthermore, acute responders in idiopathic PAH have a substantially improved prognosis compared with nonresponders [83]. As proposed by the ESC/ERS guidelines [1], a positive acute vasodilator response (inhaled nitric oxide, intravenous epoprostenol and i.v. adenosine) is defined as a ≥10 mmHg fall in mPAP to an absolute level of ≤40 mmHg accompanied by an unaltered or improved cardiac output.
The development of pulmonary oedema during acute vasodilator testing has been reported in PVOD [70, 84]. A series of 24 patients with histologically confirmed PVOD did not report any occurrence of pulmonary oedema when inhaled NO (10 ppm) was used for acute vasodilator testing and given briefly for 5–10 min [25]. Thus, very short-term administration of inhaled NO appears to be safe in PVOD. However, the absence of pulmonary oedema during acute testing does not provide any predictive information on those who will subsequently develop pulmonary oedema following initiation of targeted PAH therapy [25]. In a previous study, we reported a positive response of 12.2% in 41 PVOD patients but none of them presented with a long-term response to calcium channel blockers [81]. Importantly, PVOD patients who respond acutely to inhaled NO may develop severe pulmonary oedema rapidly after initiation of calcium channel blocker therapy [25, 81]. Given the above considerations, acute vasoreactivity testing is not routinely required in PVOD [1].
Radiographic findings
Plain radiography usually offers limited help in the differential diagnosis of PVOD unless overt pulmonary oedema is present [25, 70]. Instead, HRCT of the chest has become the mainstay imaging modality to assist with the noninvasive diagnosis PVOD. A triad of characteristic HRCT features has been described in PVOD [12, 13, 85, 86], these include: 1) mediastinal lymph node enlargement; 2) centrilobular ground-glass opacities; and 3) smooth thickening of the interlobular septa (figure 5). Data from patients with histologically proven PVOD confirmed the diagnostic utility of HRCT and demonstrated that 75% of these patients have at least two of the abovementioned HRCT abnormalities [25]. However, the corollary is that the absence or the presence of only one HRCT sign does not completely rule out the possibility of PVOD.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
High resolution computed tomography features of pulmonary veno-occlusive disease. a, b) Presence of septal lines and centrilobular ground-glass opacities. c, d) latero-aortic and subcarinal lymph node enlargement.
Günther et al. [51] documented that 61.5% of patients with systemic sclerosis-associated precapillary PH have at least two characteristic signs of PVOD on HRCT. Notably, those with at least two PVOD signs were associated with a high risk of pulmonary oedema following PAH therapy (eight out of 16 patients) and more rapid progression to death or lung transplantation. The important prognostic and management implications of these findings require further confirmation in larger cohorts of systemic sclerosis patients. Nevertheless, both histological and radiological studies support the assertion that venous involvement in systemic sclerosis-associated precapillary PH is a common occurrence.
Ventilation/perfusion lung scan
A ventilation/perfusion (V′/Q′) lung scan is recommended by guidelines as a routine investigation in the diagnostic work-up of PH in order to detect chronic thromboembolic disease, which is characterised by the presence of unmatched perfusion defects [1]. Although it has been suggested that unmatched perfusion defects may also be found in PVOD [3], a recent study demonstrated that the vast majority of V′/Q′ lung scans in patients with PVOD are in fact normal [87]. Small non-matched defects were infrequently seen in PVOD patients (7.1%) and occurred at similar rates to idiopathic PAH (10%). Therefore, a V′/Q′ lung scan should not be considered as a useful test for discriminating PVOD from PAH.
Lung function, gas exchange and exercise testing
Despite the presence of parenchymal abnormalities on HRCT, spirometry and lung volumes are typically preserved in PVOD [25]. Severe reduction in the diffusing capacity of the lung for carbon monoxide (DLCO) to less than 50% of the predicted value is common in PVOD [25], in contrast with idiopathic or heritable PAH where DLCO is relatively preserved for a long time [88]. The lower DLCO observed in PVOD could be explained by a greater reduction in capillary blood volume from a compromised pulmonary vascular bed and poorer membrane diffusion as a result of interstitial oedema. Arterial blood gas analysis typically reveals major resting hypoxaemia in PVOD compared with idiopathic or heritable PAH. Montani et al. [25] showed that mean partial pressure of arterial oxygen in PVOD patients averaged 61.3±17.3 mmHg versus 75.4±3.8 mmHg in PAH. Data on 6-min walking distance in PVOD are scarce; however, oxygen desaturation nadir during the 6-min walk test has been shown to be consistently lower in PVOD compared with PAH [19]. Thus, DLCO and arterial oxygenation parameters can be used as ancillary diagnostic tools to assist with discriminating PVOD from idiopathic PAH.
The physiological response during cardiopulmonary exercise testing in PVOD was recently reported by Laveneziana et al. [89] in a homogeneous group of heritable PVOD patients harbouring EIF2AK4 biallelic mutations. Compared with idiopathic PAH, the PVOD group had lower peak oxygen consumption, greater oxygen desaturation during exercise, greater ventilatory inefficiency, higher dead-space ventilation at peak exercise and greater dyspnoea intensity as measured by the Borg score. PAH and PVOD patients were well matched in terms of baseline anthropometric measurements, lung function and resting haemodynamics. Thus, at similar levels of haemodynamic derangement, PVOD patients have heightened sensation of dyspnoea, worse aerobic exercise capacity and greater gas exchange abnormalities.
Bronchoalveolar lavage
Due to the presence of post-capillary block, occult alveolar haemorrhage is a common histological finding in PVOD. Rabiller et al. [19] found that haemosiderin-laden macrophages were significantly increased in the BAL fluid of PVOD patients resulting in high Golde scores. Although not routinely performed in the diagnostic work-up of PH, bronchoscopy with BAL can be a useful investigation in patients with suspected PVOD, unless severe hypoxaemia is present rendering the procedure unsafe. Similar to chest HRCT features of PVOD, the absence of alveolar haemorrhage does not exclude a diagnosis of PVOD. Transbronchial biopsy should never be performed in patients with severe PH due to the unacceptably high risk of life-threatening bleeding. Finally, a recent preliminary study suggested that an increased number of haemosiderin-laden macrophages could also be detected in sputum specimens of PVOD patients [90]. The clinical utility of sputum examination as a noninvasive tool in the work-up of PVOD warrants further study.
The noninvasive diagnostic approach
Although histology remains the gold standard for a definitive diagnosis of PVOD, lung biopsy is contraindicated in the setting of clinically important PH [1]. Because histological confirmation may only be performed on post-mortem or explanted lung specimens, a noninvasive diagnostic approach is required. At the French Referral Centre, we have been using a noninvasive diagnostic strategy based on clinical features and results obtained from a battery of investigations (table 2). As detailed above, a number of salient features can help distinguish PVOD from PAH: the combination of very low DLCO, resting hypoxaemia, severe desaturation on exercise, two or more characteristic radiological signs on chest HRCT and occult alveolar haemorrhage on BAL can all be used to support a diagnosis of PVOD. Finally, genetic analysis could identify heritable PVOD in the presence of EIF2AK4 biallelic mutations. Due to the autosomal recessive transmission of heritable PVOD, consanguinity and a family history of PH in only one generation should raise the suspicion of heritable PVOD.
Comparison of key distinguishing features between pulmonary arterial hypertension (PAH) and pulmonary veno-occlusive disease (PVOD)
Natural history and prognosis of PVOD
Data on the natural history of PVOD are limited, but the disease is usually characterised by inexorable progression. Prognosis is considered to be dismal and significantly worse than other forms PAH. Survival data has been reported in two case series in the literature. In a series of 11 PVOD patients, Holcomb et al. [70] reported a mortality rate of 72% within 1 year of diagnosis. A more recent and larger series by our group found that the mean time from diagnosis to death or lung transplantation was 11.8 months, and the mean time from the first reported symptom to death or lung transplantation was 24.4 months [25]. The rapid and progressive course of PVOD requires the clinician to have a high index of suspicion in order to achieve timely diagnosis and early referral for lung transplantation.
Therapeutic options in PVOD
The past decade has witnessed a tremendous expansion of pharmacotherapeutic options for patients with PAH. Advanced therapy with “targeted PAH” drugs such as prostanoids, endothelin-1 receptor antagonists, phosphodiesterase type 5 inhibitors and soluble guanylate cyclase stimulators have significantly improved the quality of life and outlook for PAH patients [83, 91]. By contrast, randomised controlled trials are lacking in PVOD and there remains no medical therapy with proven efficacy.
General and supportive measures
Hypoxaemia should be corrected by oxygen administration to prevent further aggravation of PH from hypoxic pulmonary vasoconstriction. No outcome data on anticoagulation in PVOD exists and its use has been extrapolated from recommendations made for PAH [92]. Similar to PAH, in situ thromboses of the pulmonary microcirculation are observed in PVOD [4], supporting the biological rationale of anticoagulation. However, occult pulmonary haemorrhage is also a common finding in PVOD and anticoagulation should not be given to patients with a history of haemoptysis. Recent ESC/ERS guidelines concluded that evidence of oral anticoagulation is confined to patients with idiopathic PAH, heritable PAH and PAH due to anorexigens. No recommendation of anticoagulation has been proposed for PVOD [1].
Targeted PAH therapy
Regulatory authorities worldwide have now approved numerous drugs for treatment of PAH. Currently approved PAH drugs target one of the three major pathways involved in PAH pathogenesis: the prostacyclin, endothelin-1 and NO pathways (table 3) [83]. All PAH drugs have vasodilatory properties and also variable antiproliferative effects on the pulmonary vasculature [93]. However, data on the use of PAH therapy in PVOD are weak and conflicting. Case reports/series and anecdotal experience suggests that some PVOD patients may derive haemodynamic and functional improvements with PAH therapy, or at least a stabilisation of the disease course [70, 94–104]. However, a long-term clinical response is rarely witnessed.
Approved pulmonary arterial hypertension therapies
PAH therapies must be used with great caution in patients with PVOD and treatment should only be conducted in expert pulmonary vascular centres with experience in the management of this condition. Life-threatening pulmonary oedema is the feared complication of pulmonary vasodilatory therapy in PVOD, and can occur with any class of PAH drug [2]. Prevalence of pulmonary oedema following initiation of pulmonary vasodilators may be as high as 75% for calcium channel blockers [70] and up to 50% with targeted PAH drugs [25, 51].
The largest published experience of PAH therapy in PVOD is with the use of i.v. epoprostenol in 12 patients with severe PVOD as a bridge to transplantation [97]. Epoprostenol was given at low dose ranges (median peak dose of 13 ng·kg−1·min−1) with slow up-titration of the dose. All patients receiving epoprostenol were also concurrently listed for transplantation. High-dose loop diuretics are required to minimise the risk of pulmonary oedema. Moderate clinical and haemodynamic improvement may be observed after 3–4 months, but without long-term sustained effects. Mild clinical improvement or stabilisation of disease has also been reported with other PAH drugs such as bosentan [101–103], sildenafil [99, 100, 104] and iloprost [98].
With multiple drug classes now available to treat PAH, sequential combination therapy using a goal-oriented approach has become the standard of care for PAH [83, 105]. Increasingly, upfront combination therapy is also adopted for PAH patients, particularly if high-risk features are present [106–108]. We have reported a case of systemic sclerosis-associated PVOD where sequential combination therapy resulted in severe pulmonary oedema when sildenafil was added due to an inadequate response to bosentan monotherapy [109]. It is our opinion that all patients diagnosed with PAH should be carefully screened for the possibility of PVOD, and careful monitoring must be exercised when PAH therapies are used in cases of suspected PVOD.
Immunosuppressive therapy
The possibility of an underlying inflammatory component in the pathogenesis of PVOD has led to previous anecdotal attempts to employ immunosuppressive therapies such as glucocorticoids, cyclophosphamide and azathioprine [3]. For idiopathic PVOD, few if any reports have described clinical improvement or palliation of symptoms with immunosuppressive therapy. Similarly for patients with PAH, immunosuppression is not recommended by current guidelines [1], although there is renewed enthusiasm to explore novel and more specific targets of immunomodulation in PAH. One exception with the use of immunosuppression in PAH is in the context of systemic lupus erythematosus and mixed connective tissue disease, where glucocorticoids in combination with cyclophosphamide have a possible role and successive outcomes have been reported. In contrast, immunosuppression is not effective for systemic sclerosis-associated PAH. Given the potential harm involved, immunosuppression cannot be recommended at present for cases of idiopathic, heritable or systemic sclerosis-associated PVOD.
Lung transplantation
Lung or heart–lung transplantation remains the only definitive therapy that may offer patients with PVOD the potential for long-term survival. Given that PVOD is rapidly progressive and most patients present with advanced disease, early referral for transplantation should be considered at the time of diagnosis for eligible patients. Bilateral lung transplantation is the most widely used technique worldwide for PVOD, and post-transplant survival appears comparable to that of idiopathic PAH. A recent study reported the evolution of 49 PVOD patients listed for lung transplantation [110]. By 6 months, 22.6% of PVOD patients had been removed from the waiting list due to death, as opposed to 11% of PAH patients. After adjusting for confounding factors, diagnosis of PVOD was significantly associated with a higher risk of waiting list removal due to death compared with PAH (hazard ratio 2.05; p<0.05) [110]. These results confirmed the poor prognosis of PVOD patients and the importance of referring PVOD patients for lung transplantation early. To date, no histologically proven recurrence of PVOD after lung transplantation has been reported.
Conclusions and future directions
PVOD is an uncommon form of PH, but clinical recognition is crucial due to its dismal prognosis and nonresponsiveness to PAH therapy. Diagnosis remains challenging in clinical practice, but a high degree of diagnostic confidence is possible based on a battery of noninvasive investigations. The discovery of biallelic mutations of the EIF2AK4 gene in heritable disease will pave the way to allow a better understanding of the molecular pathogenesis of PVOD, akin to the manner in which the discovery of BMPR2 mutation in PAH has led to a major advancement of knowledge. Animal models of PVOD will enable novel therapeutic targets to be tested with eventual translation to human studies. Lung transplantation remains the current therapy of choice for eligible patients with PVOD.
Footnotes
Editorial comment in: Eur Respir J 2016; 47: 1334–1335.
Previous articles in this series: No. 1: Collard HR, Bradford WZ, Cottin V, et al. A new era in idiopathic pulmonary fibrosis: considerations for future clinical trials. Eur Respir J 2015; 46: 243–249. No. 2: Ryerson CJ, Cottin V, Brown KK, et al. Acute exacerbation of idiopathic pulmonary fibrosis: shifting the paradigm. Eur Respir J 2015; 46: 512–520. No. 3: Harari S, Torre O, Cassandro R, et al. The changing face of a rare disease: lymphangioleiomyomatosis. Eur Respir J 2015; 46: 1471–1485.
Conflict of interest: Disclosures can be found alongside the online version of this article at erj.ersjournals.com
- Received January 6, 2016.
- Accepted February 4, 2016.
- Copyright ©ERS 2016
References











Jump To
- Article
- Abstract
- Abstract
- Introduction
- PVOD in the current PH classification system
- Histological features
- Genetics of PVOD
- Risk factors and associated conditions in PVOD
- Molecular pathogenesis and animal models of PVOD
- Epidemiology
- Diagnosis of PVOD
- Natural history and prognosis of PVOD
- Therapeutic options in PVOD
- Conclusions and future directions
- Footnotes
- References
- Figures & Data
- Info & Metrics