





Abstract
Idiopathic pulmonary fibrosis (IPF) is a progressive lung disease with poor prognosis. IPF appears to be heterogeneous in pathobiology with ∼40% of IPF patients found to have elevated levels of circulating antibodies to the autoantigen type V collagen (col(V)).
Following a targeted, precision medicine approach, we conducted a phase 1 study to test the safety and explore potential efficacy of IW001, a col(V) oral immunotherapeutic developed to treat antibody-positive IPF patients. We divided 30 antibody-positive IPF patients into three cohorts for daily dosing over a 24-week period.
All patients completed treatment without serious adverse events, acute exacerbations or IPF-related hospitalisations. A decline in lung function occurred in the lowest-dose cohort that was comparable to that reported in placebo arms of published IPF trials. In contrast, the highest-dose cohort showed a trend toward stabilisation of forced vital capacity and matrix metalloproteinase 7, and a reduction in binding of C1q to anti-col(V) antibodies.
IW001 may modulate the immune response to col(V) and may represent a new therapeutic for col(V)- reactive IPF patients.
Abstract
IW001 is safe, and improves biomarkers and lung function in IPF patients with type V collagen antibodies http://ow.ly/LirbO
Introduction
Idiopathic pulmonary fibrosis (IPF) is a chronic interstitial lung disease of unknown aetiology. Repetitive, widespread injury to the alveolar epithelium and aberrant wound repair processes appear to be the driving forces behind IPF pathogenesis [1], which is marked by uncontrolled deposition of extracellular matrix in the lung, resulting in the loss of pulmonary function. The prognosis is very poor, with a median survival of ∼3 years following diagnosis [1, 2].
The pathophysiology of IPF is unknown. Multiple dysregulated processes, including coagulation, inflammation, adaptive immunity and fibroblast activity are involved in the pathogenesis or perpetuation of IPF [3–5]. Research has not yet determined how or if the relative importance of individual pathways varies among individuals with IPF [6]. The ability to classify patient subsets would allow implementation of a precision medicine approach to treat affected patients [7].
The role of humoral autoimmunity in IPF is an emerging subject of investigation. Autoantibodies are present in the plasma of >80% of patients with IPF, including specific autoantibodies to heat-shock protein 70 and K-α1 tubulin [8, 9]. Autoreactive T-cells against lung antigens were also detected in the same studies, supporting the idea that an ongoing adaptive immune response against autoantigens may play an important role in the disease progression for some IPF patients. Previous studies have shown that 40–60% of IPF patients may display an autoimmune response against type V collagen (col(V)) [10, 11].
Col(V) is a minor collagen found primarily within the fibrils of the major lung collagen, type I. Col(V) performs many functions, including providing elasticity within the lung architecture [10], organising and maintaining the spatial configuration of collagen fibres [12], controlling the formation the fibre mesh and the diameters of other collagen fibres, and linking stromal collagen to the basement membrane. Lung injury may lead to the exposure of this normally sequestered antigen, rendering it available for activation of an autoimmune response [13]. A sustained autoimmune response may lead to abnormal lung remodelling, as evidenced by lung biopsies of IPF patients showing that increased levels of col(V) correlate with fibrosis and disease progression, and are a predictor of survival [12]. Vittal et al. [10] reported that col(V) was highly overexpressed in the IPF lung. In addition, pre-clinical studies have confirmed that anti-col(V) autoimmunity can result in fibrotic changes in the lung [11, 14].
Oral immunotherapy (OIT) induces clinical desensitisation of food allergies and autoimmune diseases. Pre-clinical animal studies have shown that oral or nasal administration of purified col(V) can suppress both humoral and cell-mediated immune responses against it, resulting in the preservation of lung tissue in fibrosis related to lung transplant rejection [15, 16] and in bleomycin-induced pulmonary fibrosis [10].
In this phase 1 clinical trial, we hypothesised that OIT with col(V) (IW001) in IPF patients who were anti-col(V) antibody (Ab)+ would be a safe and effective means to induce an antigen-specific alteration of the immune response and stabilise lung function. This open-label, multicentre study was designed to evaluate the safety, tolerability, and biological and clinical effects of three different doses of IW001 in IPF patients who were anti-col(V) Ab+.
Materials and methods
Study design
The clinical trial design is depicted in figure 1. A total of 85 IPF patients (diagnosed according to American Thoracic Society (ATS)/European Respiratory Society (ERS)/Japanese Respiratory Society (JRS)/Asociación Latinoamericana de Tórax (ALAT) criteria [17]) from 11 clinical sites were screened for anti-col(V) antibodies. 34 patients were anti-col(V) Ab+ and of these, 30 patients met all entry criteria and were enrolled into the study. Patients were treated in three dose cohorts: 0.1, 0.5 and 1.0 mg daily of IW001 (oral solution of bovine col(V)) for 24 weeks. Patients were enrolled sequentially into the study (lowest dose to highest). Patients in cohort 1 received treatment with IW001 at 0.1 mg per day. When enrolment into this low-dose cohort was complete and all cohort 1 patients had been treated for ≥30 days, cohort 2 was enrolled. Patients in cohort 2 were given 0.5 mg per day. When enrolment to this mid-dose cohort was complete and all cohort 2 patients had been treated for ≥30 days, cohort 3 was enrolled. Patients in cohort 3 were given 1.0 mg per day. At study enrolment, and months 3 and 6, patients underwent a physical examination and pulmonary function studies. Blood was collected monthly for biomarkers.
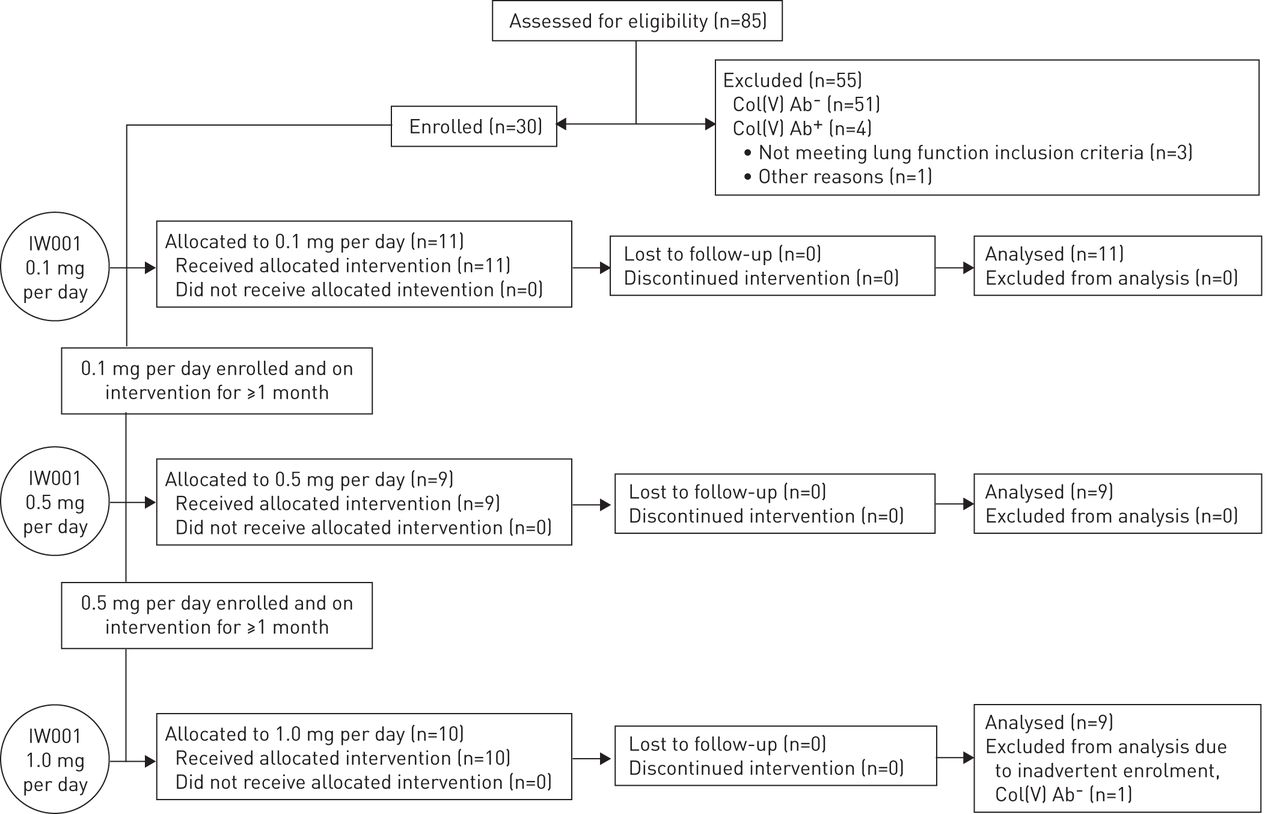
Trial design. Patients in cohort 1 received treatment with IW001 at 0.1 mg per day orally for 24 weeks. When enrolment to this cohort was complete and all cohort 1 patients had been treated for ≥30 days, cohort 2 was enrolled. Patients in cohort 2 were given IW001 at 0.5 mg per day orally for 24 weeks. When enrolment to this cohort was complete and all cohort 2 patients had been treated for ≥30 days, cohort 3 was enrolled. Patients in cohort 3 were given IW001 at 1.0 mg per day orally for 24 weeks. Col(V): type V collagen; Ab: antibody.
Key inclusion criteria for this clinical trial included: age 35–75 years (inclusive); diagnosis of IPF (ATS/ERS/JRS/ALAT criteria [17]) within the previous 48 months; forced vital capacity (FVC) ≥50% of predicted; diffusing capacity of the lung for carbon monoxide >35% of predicted; and positive for IgG antibodies against col(V) (Ab+). Key exclusion criteria included: use of systemic corticosteroids and/or immunosuppressives; allergies to bovine products; recent smoking; any active infection; unstable cardiovascular disease; or any condition that might interfere with absorption of IW001.
Anti-col(V) Ab detection
Antibodies to col(V) were detected in patients' plasma using a patented flow cytometry bead assay (ImmuneWorks, Indianapolis, IN, USA) [18]. A floating cut-off was determined by using serum from 66 adult, nonsmoking normal volunteers, following an immunogenicity white paper [19].
Peripheral blood biomarker analyses
C1q
The quantity of C1q bound to anti-col(V) antibodies in serum was determined by ELISA. In a 96-well plate, patient-derived heat-inactivated serum was added to four wells coated with IW001 (col(V)). After incubation and washing, a known amount of C1q (Sigma-Aldrich, St Louis, MO, USA) was added to two wells, while the remaining wells were treated with buffer. After detection with horseradish peroxidase-conjugated anti-C1q detection Ab (MyBiosource, San Diego, CA, USA), the optical densities (ODs) of the wells not treated with C1q were subtracted from the OD measured in the wells treated with C1q, in order to determine the change in C1q binding.
Matrix metalloproteinase 7
Plasma matrix metalloproteinase (MMP)7 levels were measured using the Quantikine MMP7 ELISA Kit (R&D Systems, Minneapolis, MN, USA), according to the manufacturer's protocol.
Serum albumin
Serum albumin assays were performed by Covance Central Laboratory (Indianapolis, IN, USA) using a Modular Analyzer (Roche Diagnostics, Indianapolis, IN, USA). At the reaction pH, bromocresol purple binds selectively to albumin. The intensity of the colour was measured photometrically and is proportional to the total amount of albumin present.
Statistical analyses
Data are presented as mean±sem unless otherwise specified. Statistical analyses were performed with Prism software (GraphPad Software, Inc., La Jolla, CA, USA).
Nonparametric tests were used in regression analyses as well as when comparing outcomes from the 0.1 mg per day with the 1.0 mg per day cohorts. In all cases, p-values <0.05 were considered statistically significant. One patient was enrolled in the high-dose group (1 mg per day) whose anti-col(V) Ab result was later discovered to be negative (laboratory handling error). Consequently, data from this patient were included in the safety analyses but excluded from the efficacy analyses. All patients completed the study, so no handling of missing data was conducted for this publication.
Results
Phase 1 clinical trial patient demographics and relationship to col(V) Ab status
34 (40%) out of 85 patients screened were col(V) Ab+. Of these, 30 patients were enrolled and table 1 shows baseline patient demographics. One patient was enrolled in the high dose group (1 mg per day) whose anti-col(V) Ab result was later discovered to be negative (laboratory handling error). Consequently, data from this patient are included in the safety analyses but excluded from the efficacy analyses.
Summary of the patient demographics
IW001 safety profile
The primary end-point of the trial was safety/tolerability. A total of 26 adverse events deemed potentially related to IW001 were reported in 11 patients. All were rated as either mild or moderate. These are summarised in table 2. The majority of the adverse events were gastrointestinal disorders, including diarrhoea and soft stools. No acute exacerbations of IPF or hospitalisations were observed. 100% of the patients enrolled completed the trial; no patients discontinued early for any reason.
Summary of individual adverse events potentially related to IW001
Effect of IW001 on lung function
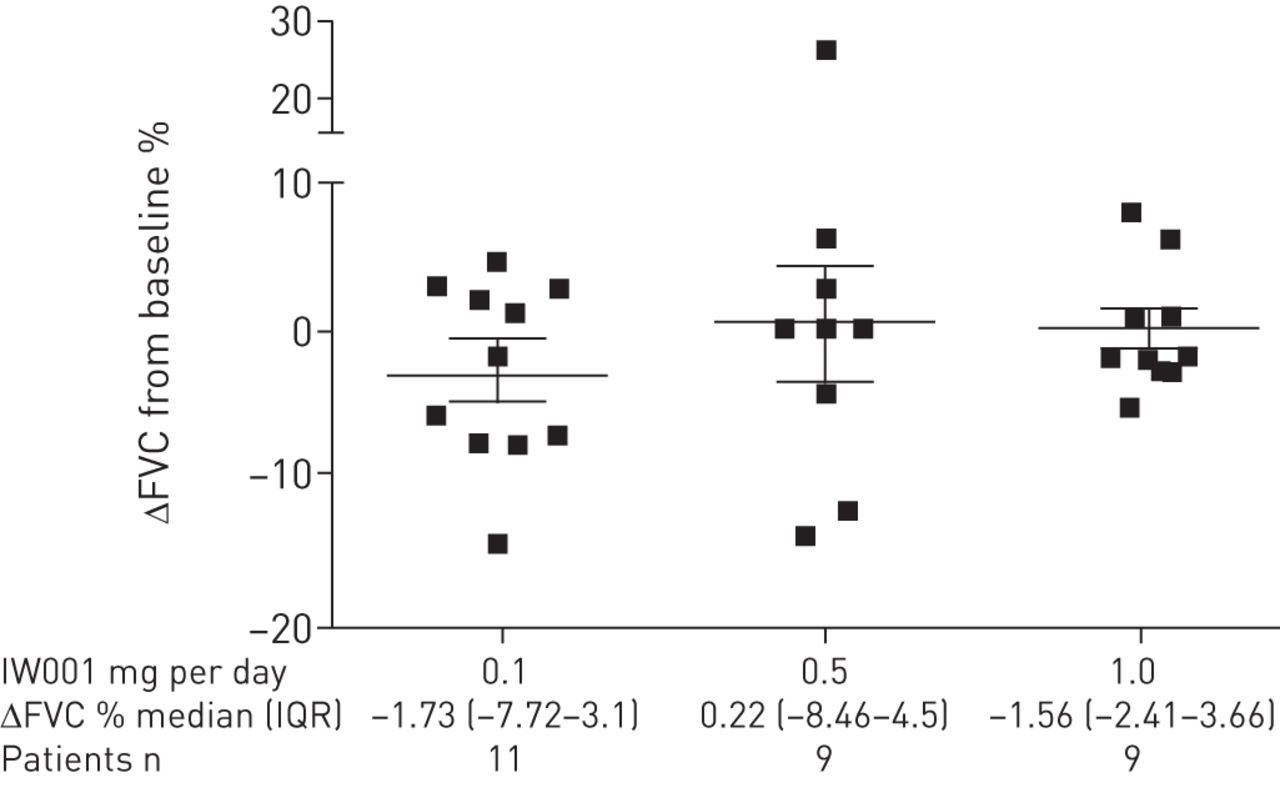
A secondary objective of this phase 1 clinical trial was to determine changes in lung function. The results showed that there was a difference in the mean change in FVC among the three doses after 24 weeks of treatment with IW001 (fig. 2), calculated as (FVC at 24 weeks in litres−FVC at baseline in litres)/FVC at baseline in litres. The patients in the 0.1 mg per day cohort showed a mean±sem change from baseline of −2.75±1.87% over the 24 week treatment period. In contrast, the patients in the 0.5 mg per day cohort showed a mean change of 0.57±3.90% and the patients in the highest drug dose cohort (1.0 mg per day) showed a mean change from baseline FVC of 0.33±1.44%.

Change in (Δ) forced vital capacity (FVC) from baseline after 24 weeks of treatment with IW001 indicates a potential reduction of lung function decline. Patients' FVCs were measured at enrolment and after 24 weeks of treatment with IW001. The percentage change from baseline was calculated as ΔFVC=(FVC at 24 weeks in litres−FVC at baseline in litres)/FVC at baseline in litres. Data in the graph are presented as mean±sem for each cohort. IQR: interquartile range.
As individual IPF patients may experience significant reductions in lung function as measured by FVC, which could skew the mean change seen for the group of patients, we also conducted a categorical analysis of the changes in FVC % predicted on an individual basis for this trial. Using an absolute decline in FVC % predicted of 5% for a 6-month study as a predictor of decreased median survival time, as defined by Zappala et al. [20], table 3 shows that four of the 11 patients enrolled in the lowest drug dose cohort showed a decline of ≥5% in their FVC over the course of 24 weeks. In contrast, only two out of nine patients in the intermediate drug dose cohort (0.5 mg per day) showed a similar decline in FVC. No patient in the highest drug dose cohort (1.0 mg per day) showed a decline of ≥5% in FVC.
Categorical analysis of forced vital capacity (FVC) decline after 24 weeks of treatment indicate that the 1.0 mg dose of IW001 may stabilise lung function
Effect of IW001 on biomarkers
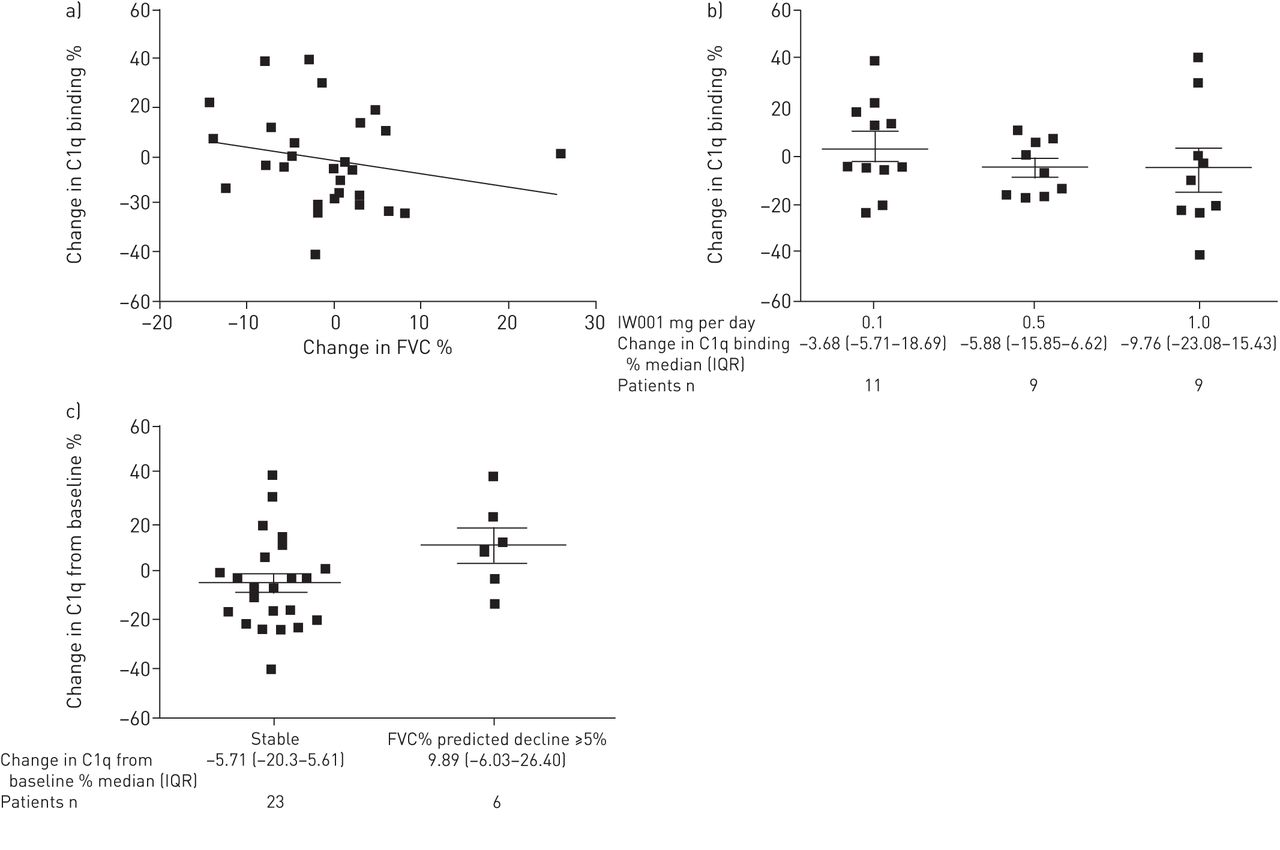
C1q was chosen as a biomarker of the immunological effect of IW001, as a decrease in C1q binding to anti-col(V) antibodies would indicate an IW001-induced effect on underlying anti-col(V) Ab binding and activity. C1q binding to anti-col(V) antibodies was assessed and correlated with responses to IW001 treatment. Figure 3a shows the correlation between percentage change in C1q binding and that in FVC; figure 3b shows that C1q binding to anti-col(V) antibodies increased slightly in the patients receiving 0.1 mg per day of IW001 while patients receiving 0.5 mg per day and 1.0 mg per day had dose-dependent decreases in C1q binding.

a) Correlation between percentage change from baseline in forced vital capacity (FVC) and C1q binding. Patient blood samples were collected at initiation of the trial and after 24 weeks of treatment with IW001. The percentage change from baseline in C1q bound to anti-type V collagen (col(V)) antibodies following 24 weeks of treatment was plotted against the percentage change from baseline in FVC. The percentage change from baseline was calculated as (FVC at 24 weeks in litres−FVC at baseline in litres)/FVC at baseline in in litres, and (C1q OS at 24 weeks–C1q OD at baseline)/C1Q OD at baseline. n=29 patients. Spearman’ nonparametric correlation: r= −0.2729, p=0.1520. b) C1q binding to anti-col(V) antibodies. The percentage change from baseline in C1q bound to anti-col(V) antibodies from patients' heat inactivated sera following 24 weeks of treatment was calculated as (C1q OD at 24 weeks–C1q OD at baseline)/C1q OD at baseline and is reported for each cohort. Data in the graph are presented as mean±sem for each cohort. Comparison between lower and higher dose: Mann–Whitney nonparametric test p=0.4033. c) Correlation between FVC and C1q binding. Patients whose FVC % predicted decreased by ≥5% (n=6) over the course of 24 weeks of treatment were compared with patients whose FVC appeared more stable (n=23) for the changes in binding of C1q to their anti-col(V) antibodies after 24 weeks of treatment. The percentage change from baseline of C1q binding to anti-col(V) antibody after 24 weeks of treatment is reported for each group, calculated as FVC % predicted at 24 weeks−FVC % predicted at baseline. Data in the graph presented as mean±sem for each group. Comparison between stable patients and patients with declining FVC % predicted ≥5%: Mann–Whitney nonparametric test p=0.071. OD: optical density; IQR: interquartile range.
We analysed the effect of C1q binding on anti-col(V) antibodies among those patients from all three cohorts who showed a decline of ≥5% (absolute) in FVC % predicted over the course of treatment with IW001 (n=6), and compared those results with the patients who displayed stable lung function (n=23). Those patients with a significant decrease in FVC had higher level of C1q binding on anti-col(V) antibodies after 24 weeks, compared with those with a stable lung function and whose C1q binding activity decreased over the course of treatment (fig. 3c).
MMP7 has been shown to be a biomarker that is predictive of IPF progression [21–23]. Plasma MMP7 levels were determined at baseline and week 24. Compared with baseline, MMP7 increased by 585.5±463.3 pg·mL−1 in the 0.1 mg per day cohort by 24 weeks (fig. 4a). In contrast, MMP7 increased by 1416±1252 pg·mL−1 in the 0.5 mg per day cohort. Patients in the cohort receiving the 1.0 mg per day dose showed a mean decrease in serum MMP7 levels over the same treatment period (−65.33±492.2 pg·mL−1).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
a) Plasma matrix metalloproteinase (MMP)7 levels. Blood samples were collected at the initiation of the trial and after 24 weeks of treatment with IW001. Absolute changes in plasma levels of MMP7 following 24 weeks of treatment are reported for each cohort. Data in the graph are presented as mean±sem for each cohort. Comparison between lower and higher dose: Mann–Whitney nonparametric test, p=0.8197. b) Serum albumin levels. Blood samples were collected at the initiation of the trial and after 24 weeks of treatment with IW001. Absolute changes in serum albumin levels following 24 weeks of treatment are reported for each cohort and were calculated as albumin at 24 weeks in g·dL−1−albumin at baseline in g·dL−1. Data in the graph are presented as mean±sem for each cohort. Comparison between lower and higher dose: Mann–Whitney nonparametric test, p=0.0413. IQR: interquartile range. *: p<0.05.
To examine the effect of IW001 on the overall wellbeing of treated patients, serum albumin levels were measured, as recent studies indicated an inverse correlation with mortality in IPF patients [22, 24]. Serum albumin decreased in the patients receiving the lowest does of IW001 (0.1 mg per day) over the course of the 24-week treatment period, whereas the levels were slightly increased at the 0.5 mg per day dose. In contrast, patients on the 1.0 mg per day dose showed a statistically significant increase in serum albumin levels, as compared with cohort 1 (fig. 4b).
Discussion
This 6-month, open-label clinical trial of IW001 OIT in col(V) Ab+ IPF patients shows a number of interesting observations. The prevalence of anti-col(V) antibodies is ∼40% in IPF patients. col(V) OIT was safe and well tolerated in all patients, and appears to lead to a dose-dependent trend in stabilisation of lung function and circulating MMP7 levels, and decreases in C1q binding to anti-col(V) antibodies. Finally, IW001 induced dose-dependent increases in serum albumin, a protein in which decreased serum levels have been linked to decreased survival in IPF patients.
IPF affects ∼500 000 people in the USA and Europe [25], and its incidence appears to be increasing, as reported in multiple countries through registries and other epidemiological vehicles [6, 26]. This underscores the need for effective treatments that are safe for chronic use in an older patient population; however, that goal remains elusive. Lung transplantation, while sometimes effective, is an option only for certain IPF patients [1]. Altering the roles of coagulation, inflammation, oxidative stress and excessive deposition of extracellular matrix by fibroblasts in IPF have been tested in numerous clinical trials. Given the diverse pathophysiology that constitutes IPF, highly targeted therapies may be inadequate for making a detectable difference in disease progression [6].
Col(V) is a minor collagen normally sequestered within the lung interstitium and, therefore, hidden from the immune system. Lung remodelling, a characteristic feature of IPF, leads to col(V) exposure and overexpression, which in turn is associated with the development of anti-col(V) immunity, indicating an autoimmune aspect to IPF pathogenesis. Specifically, up to 60% of IPF patients have anti-col(V)-reactive T-cells [27] and, as shown in the current study and a report by Vittal et al. [10], with >40% of 155 IPF patients exhibiting specific systemic Ab responses.
One approach that, heretofore, was absent from clinical trial activity was testing the possibility that an altered adaptive immune response may be important in regulating the fibrotic response in these patients. Because our data indicate that ≥40% of IPF patients have an ongoing autoimmune response against col(V), this autoantigen may not only play a role in disease progression for this subset of patients, but suggests that therapeutically altering the autoimmune response may lessen the fibrotic response and stabilise lung function. Other autoantibodies, including those to heat-shock protein 70, and autoreactive T-cells against lung antigens exist in the plasma of >80% of patients with IPF [8, 9]. Collectively, this supports the idea that an ongoing adaptive immune response against autoantigens may play an important role in disease progression for IPF patients who test positive for anti-col(V) antibodies.
Because IgG isotypes vary in their ability to bind C1q and activate the complement cascade, the decreased C1q binding seen in patients treated with the highest dose of IW001 suggests changes in the pool of anti-col(V) antibodies in these patients. The binding affinity for C1q depends on an amino acid sequence in the heavy chain of the antibodies [28]. Decreased C1q binding observed in the current study could suggest a change in the amino acid sequence of the Ab, causing an increase in a type of Ab that has a lower affinity for binding complement, as well as the previously resident anti-col(V) Ab. Other possibilities of how this change in C1q binding can occur could range from selective loss of complement-binding antibodies to altered isotype switching in B-cells and is a topic worthy of further investigation. However, decreases in anti-col(V) Ab post-treatment did not account for lower C1q binding, as anti-col(V) Ab levels largely remained unchanged during the trial, and those patients who displayed an increase in anti-col(V) Ab had the greatest decline in C1q binding (data not shown). Furthermore, all four IgG subtypes were present in all treatment groups. The mechanism of this altered binding is the subject of ongoing investigation.
OIT via the administration of the autoantigen to the gut has been shown to suppress both humoral and cell-mediated immune responses [29, 30]. This may be due to the activation of regulatory T-cells (Tregs), which sustain immunological self-tolerance by blocking the response of effector T-cells activated against the same autoantigen. In this clinical trial, we did not observe uniform changes in Tregs (CD4+CD25+FoxP3+ T-cells) in peripheral blood (data not shown). However, we did observe an overall increase in anti-col(V) Ab in the highest-dose cohort, which was the group with stabilised lung function, decline in MMP7, increased serum albumin and decline in C1q binding. It is interesting to speculate that the beneficial effect of IW001 reported in the current study could be linked to induction of inhibitory antibodies that block anti-col(V) Ab immune complex mediated activation of col(V)-reactive T-cells, a mechanism that has been invoked in IgE–antigen complex in successful trials of peanut OIT [31]. Future trials will investigate the mechanism of IW001-induced OIT.
Wound healing and tissue remodelling also involve the production of MMPs and increased MMP7 levels have been correlated with increased disease severity in IPF patients [23]. Thus, our data showing a trend toward stable MMP7 levels in patients receiving the highest tested dose of IW001, as well as a correlation between lower MMP7 levels and stabilised FVC after 24 weeks of treatment, support the idea that IW001 may alter disease pathology and the ongoing fibrotic response of this disease. It should be noted that the increase in MMP7 in the 0.5-mg cohort was driven mainly by one patient and direct evidence for the MMP7 stabilisation is that the 75th centile for cohort 3 was 710 pg·mL−1 versus 1500 pg·mL−1 for cohort 1.
Serum albumin has been reported recently to be associated with mortality in IPF patients. Specifically, declines in serum albumin were linked to increased mortality in these patients [22, 24]. While serum albumin may not be considered a biomarker, these recent studies suggest that therapeutics that are associated with increases or maintenance of albumin may reflect beneficial effects in IPF patients. Indeed, data in the current study showed IW001 induced dose-dependent effects on serum albumin, with the highest cohort revealing an increase in albumin levels at the completion of the trial. This same cohort showed desirable trends in FVC and biomarkers.
A potential limitation of the current study is the lack of a designated placebo group. However, lung function testing showed that patients taking the lowest dose (0.1 mg per day) displayed a similar mean decline in lung function as reported in the placebo arms of two major IPF clinical trials. In fact, placebo groups in those studies of ∼165 patients each resulted in a decline of observed FVC by week 24 of −80.7 mL and −92.1 mL respectively [32, 33], while our lower dose declined −85.4 mL. As this was a phase 1 trial, the total number of patients in enrolled could be considered low, and each end-point, when examined individually, could be of limited interpretative value. However, collective examination of the data showed all end-points pointing in the direction of benefit to patients in the high-dose (1 mg per day) group, which include positive trends in all biomarkers, serum albumin and lung function.
While this study was not designed to evaluate disease progression or mortality, the collective changes in our series of surrogate markers for these clinical outcomes suggest that treatment with IW001 can alter the underlying biology of this disease in these patients. Future proof-of-concept clinical trials that test IW001 for a longer time frame in a larger number of IPF patients against a placebo arm may validate this approach as an effective treatment for a significant subset of IPF patients. Additional studies to elucidate the roles of C1q binding and MMP7 levels during the treatment of IPF patients with IW001 may advance our knowledge of how to preserve lung function in these patients.
In conclusion, this is the first clinical trial to treat a prospectively defined subset of IPF patients, specifically those who were anti-col(V) Ab+, with an OIT approach using col(V). IW001 appears to be safe and well-tolerated, shows evidence of biological activity based on reduced binding of C1q to anti-col(V) Ab, and may have a beneficial impact on disease progression based on trends in FVC. Further study with col(V) OIT is warranted to confirm the therapeutic effect of IW001in IPF patients.
Acknowledgements
Thanks to Lana Christian of CreateWrite Inc. (Indianapolis, IN, USA) for her expert writing and editing assistance, and to Rocco Brunelle of B2S Consulting (Indianapolis, IN, USA) for his expert assistance in statistics. This study was made possible by the efforts of additional clinical trial investigators: Michael Busk, Mark Farber (both Indiana University School of Medicine, Indianapolis, IN, USA), Mary Strek (University of Chicago School of Medicine, Chicago, IL, USA), James Allen (Ohio State University School of Medicine, Columbus, OH, USA), Jesse Roman (University of Louisville School of Medicine, Louisville, KY, USA), Steven Sahn (Medical University of South Carolina, Charleston, SC, USA), Joao de Andrade (University of Alabama at Birmingham School of Medicine, Birmingham, AL, USA), Yolanda Mageto (University of Vermont, Burlington, VT, USA), Marilyn Glassberg (University of Miami School of Medicine, Miami, FL, USA), Lisa Lancaster (Vanderbilt University School of Medicine, Nashville, TN,USA) and Sean Studer (Newark Beth Israel Medical Center, Newark, NJ, USA).
Footnotes
For editorial comment see Eur Respir J 2015; 45: 1218–1220 [10.1183/09031936.00237114].
Support statement: This study was funded by ImmuneWorks, Inc. (Indianapolis, IN, USA).
Clinical trial: This study is registered at www.clinicaltrials.gov with identifier number NCT00873860.
Conflicts of interest: Disclosures can be found alongside the online version of this article at erj.ersjournals.com
- Received June 10, 2014.
- Accepted October 31, 2014.
- Copyright ©ERS 2015
References










