





Abstract
Small-scale clinical trials show that treatment of cystic fibrosis (CF) patients with ibuprofen, a nonsteroidal anti-inflammatory drug, improves the symptoms of CF and slows down the decline of lung function. Paradoxically, ibuprofen inhibits ligand-stimulated CF transmembrance conductance regulator (CFTR) activity. The aim of the present study was to investigate the effects of ibuprofen on CFTR function under different conditions.
Patch-clamp recordings were performed in two lines of human airway epithelial cells: IB3-8-3-7 cells, which express wild-type CFTR; and IB3-1 cells, which express the variant CFTR with deletion of phenylalanine 580 (ΔF580CFTR).
Addition of ibuprofen to the extracellular solution caused a rapid inhibition of CFTR activity in IB3-8-3-7 cells in the presence of a high intracellular concentration of cAMP, whereas ibuprofen enhanced the CFTR conductance at low levels of cAMP. Introducing ibuprofen into the interior of cells occluded the enhancing effect of ibuprofen. Notably, the variant CFTR-mediated conductance was detected in IB3-1 cells treated with myoinositol and was enhanced by ibuprofen at endogenous levels of cAMP.
In summary, nonsteroidal anti-inflammatory drugs increase the function of both wild-type cystic fibrosis transmembrane conductance regulator and the phenylalanine 580 deletion in cultured human airway epithelial cells at endogenous levels of cAMP.
Cystic fibrosis (CF) is one of the most common fatal genetic diseases and is caused by mutations in the gene encoding the CF transmembrane conductance regulator (CFTR), a cAMP-gated Cl- channel. The dysfunction of variant CFTR results in a reduction in Cl- secretion from various epithelial cells, which, in turn, leads to a reduction of the volume of airway surface liquid and, as a consequence, mucus adhesion, inflammation and bacterial biofilm formation occur in the lung. Although CF is associated with altered epithelial function in multiple visceral organs including the lung, intestine and pancreas, the pulmonary complications, such as chronic inflammation and mucous plugging, are the major causes of morbidity and mortality in this disease 1. In this regard, pharmacological management is still the major approach for dealing with pulmonary complications of CF patients 1.
CF is primarily caused by the dysfunction of variant CFTR in epithelial cells of affected organs; thus, enhancing or restoring the function of variant CFTR in CF patients should be the principle aim of CF treatment. Small-scale clinical studies have shown that ibuprofen, a nonsteroidal anti-inflammatory drug (NSAID), not only improves the symptoms but also preserves lung function in CF patients 2, when the gastrointestinal side-effects of this medication are carefully managed. The benefit of ibuprofen treatment has been largely attributed to its anti-inflammatory action. A previous study has shown that ibuprofen inhibits cAMP-activated CFTR currents in epithelial cells by an unknown mechanism 3. This inhibitory action on the CFTR would paradoxically be anticipated to exacerbate the symptoms of CF. Bearing this concern in mind, the effects of NSAIDs, specifically ibuprofen, on the function of wild-type and the phenylalanine 508 deletion variant CFTR (ΔF580CFTR) were re-examined.
EXPERIMENTAL PROCEDURES
Cell culture
IB3-1 cells 4 were originally derived from airway epithelial cells of a CF subject who expressed ΔF580CFTR 5. By generating a doxycycline-controlled expression system in IB3-1 cells, a new cell line, named IB3-8-3-7, which expresses wild-type CFTR 6, was created. IB3-1 cells and IB3-8-3-7 cells were cultured in LHC-8 medium (Invitrogen, Carlsbad, CA, USA) supplemented with 5% foetal bovine serum (Invitrogen) and 150 μg·mL−1 hygromycin B (Sigma, St. Louis, MO, USA). Inducible CFTR expression in IB3-8-3-7 cells was triggered by the addition of doxycycline (1 µg·mL−1), and the expression of CFTR was confirmed by real-time PCR 6 and immunocytochemistry assay. IB3-8-3-7 cells were used after 18–21 h of treatment with doxycycline. Some IB3-1 cells were treated with 3 mmol·L−1 myoinositol (Sigma) and used after 18–21 h of treatment with myoinositol.
Heterologous expression of γ-aminobutyric acid receptors
IB3-8-3-7 cells were transiently transfected with cDNAs encoding human A-type, γ-aminobutyric acid (GABA) receptors (GABAAR)-α1, -β2, and -γ2L subunit isoforms (1:1:1 ratio) by using Lipofectamine 2000 (Invitrogen). All cDNAs were subcloned into the mammalian expression vectors pCDM8 for heterologous expression. Transfected cells were grown and incubated for 24–48 h before use.
Patch-clamp recordings
Perforated or conventional whole-cell patch-clamp recordings were performed in IB3-1 and IB3-8-37 cells using an Axopatch-1D amplifier (Axon Instruments, Foster City, CA, USA). The perforated-patch technique was employed to avoid disturbing cell intracellular ion concentrations and metabolism. Therefore, the endogenous membrane voltage (Vm) of the cells could be detected. The conventional whole-cell recordings were used to manipulate intracellular concentrations of ions and cAMP, and to deliver certain test compounds into the cells. Before being used for recordings, the cells were rinsed with and bathed in the standard extracellular solution (ECS) containing: 145 mmol·L−1 NaCl, 1.3 mmol·L−1 CaCl2, 5.4 mmol·L−1 KCl, 25 mmol·L−1 HEPES and 10 mM glucose (kept at 35°C). Unless specifically described otherwise in the Results section, the standard intracellular solution (ICS) for whole-cell recording consisted of: 150 mmol·L−1 KCl, 10 mmol·L−1 HEPES, 2 mmol·L−1 MgCl2, 1 mmol·L−1 CaCl2 and 0.005 mmol·L−1 K2ATP, with or without added cAMP. ICS for perforated-patch recording consisted of: 150 mmol·L−1 KCl, 10 mmol·L−1 HEPES, 2 mmol·L−1 MgCl2, 1 mmol·L−1 CaCl2 and the pore-forming agent gramicidin (50 µg·mL−1; Sigma) 7. The pH of ECS and ICS was adjusted to 7.4 and 7.3, respectively, and the osmolarity was corrected to a range of 310–315 mOsm. Patch electrodes (3–5 MΩ) were constructed from thin-walled glass (1.5-mm diameter; World Precision Instruments, Sarasota, FL, USA) using a two-stage puller (PP-830; Narishige, East Meadow, NY, USA).
Perforated-patch recordings started under voltage-clamp mode. Membrane perforation was observed as a constant decrease in serial resistance after electrode seal. In most recordings, the resistance declined to a value ranging 30–35 MΩ within 5–8 min after the seal, and then the resistance could be stable for ∼40–80 min. In order to monitor a possible formation of whole-cell configuration, the serial resistance was examined approximately every 5 min during the recording. Whole-cell recordings were carried out as previously described 8, 9.
The Vm of the cell was measured using current-clamp technique. Under voltage-clamp mode, transmembrane conductance of the cell was determined by hyperpolarising or depolarising voltage steps or by a voltage ramp (a steady voltage-change from -100 to 100 mV within 1.5 s). In accordance with the experimental purpose, testing drugs were added to the ICS or ECS. The control ECS and extracellularly used drug(s) were applied to the cell using a computer-controlled two-barrel perfusion system (SF-77B; Warner Instruments, Hamden, CT, USA). Testing drugs were added to the ECS at 8–12 min after whole-cell configuration, at which the amplitude of currents was steady.
Data analysis
All the recorded electrical signals were digitised, filtered (1 kHz band pass filter) and acquired online using Clampex software and analysed offline using Clampfit software (both Axon Instruments). Data are presented as mean±sem. Means were compared with unpaired or paired t-tests, where appropriate. A p-value <0.05 was considered as significant.
RESULTS
Electrophysiological characterisation of wild-type CFTR expressed in human airway epithelial cells
The doxycycline-induced expression of wild-type CFTR in IB3-8-3-7 cells 6 was determined by immunocytochemistry (fig. 1⇓). Perforated-patch recordings revealed that the Vm of untreated IB3-8-3-7 cells was -11.6±3.1 mV, whereas the Vm of doxycycline-treated cells was -32.2±3.0 mV (fig. 2a⇓), which is close to the Vm of primary human airway epithelial cells 10. It is likely that this change of Vm was due to the suppression of endogenous epithelial sodium channels (ENaC), which are associated with increased expression of CFTR 11, 12. CFTR activity is regulated by intracellular cAMP, and the concentration of endogenous cAMP is controlled by adenylate cyclase (AC) 13. Applications of the AC activator forskolin (3 µmol·L−1) induced a substantial and long-lasting depolarisation (figs 2a⇓ and b). This depolarisation is consistent with a cAMP-induced CFTR-mediated Cl- efflux.

Immunocytochemical staining of cystic fibrosis transmembrane conductance regulator (CFTR) in a) control IB3-8-3-7 cells and b) cells treated with doxycycline. Doxycycline-treated cells are CFTR-immunopositive. Scale bars = 25μm.

a) Membrane voltage (Vm) of IB3-8-3-7 cells measured by perforated-patch current-clamp recording: in the absence (-; -11.6±3.1 mV; n = 6) and presence (+; -32.2±3.0 mV; n = 9) of doxycycline; and in doxycycline-treated IB3-8-3-7 cells before (-; -30.4±3.9 mV) and after (+; -15.6±4.9 mV; n = 5) addition of 3 μM forskolin. Data are presented as mean±sem. b) Under perforated-patch current-clamp conditions, brief addition of 3 μM forskolin (□), to the extracellular solution, induced a long-lasting depolarisation. - - - : baseline of -30 mV. *: p<0.05.
Whole-cell recordings were then performed with or without cAMP (100 µmol·L−1) in the ICS (fig. 3a⇓ and b). Every cell (at -60 mV; n = 16) responded with a slowly developing inward current in the presence of cAMP (fig. 3b⇓). This current gradually increased to reach a maximal level at 4–6 min after whole-cell configuration and then steadily declined, reaching a stable value 8–10 min after. By contrast, no current was recorded in the absence of cAMP (fig. 3a⇓). To explore whether this current was mediated by a Cl- conductance, Cl- in the ICS was replaced with gluconate, a membrane-impermeable anion. Under this condition, cells generated outward currents when held at 0 mV (fig. 3c⇓). If Cl- in the ECS was also replaced with gluconate, the outward current gradually decreased to baseline (fig. 3c⇓). This result confirms that the cAMP-induced current is mediated by a Cl- conductance. The transmembrane conductance was also examined by increasing Vm in a stepwise manner (from -100 to 100 mV; fig. 4⇓). No conductance was detected in the absence of cAMP (fig. 4a⇓), whereas a voltage-independent conductance was observed when cAMP (100 µmol·L−1) was included in the ICS (fig. 4b⇓ and d). In addition, when Vm was held below 0 mV, this conductance was strongly inhibited by glybenclamide (100 µmol·L−1), a CFTR blocker 14 (fig. 4c⇓ and d). Taken together, these data demonstrate that the doxycycline-treated IB3-8-3-7 cells express functional CFTR channels.

Typical transmembrane currents in doxycycline-treated IB3-8-3-7 cells (clamped at -60 mV) in a) the absence or b) the presence of 100 μM intracellular cAMP. c) Example trace shows the direction switch of transmembrane current in response to gluconate rather than Cl- in the extracellular solution; the cell was voltage clamped at 0 mV and the intracellular solution contained gluconate rather than Cl-, throughout. - - -: basal current of 0 A.

a) In the absence of intracellular cAMP, stepwise increase of membrane voltage (Vm) from -100 to 100 mV did not reveal transmembrane conductance in doxycycline-treated IB3-8-3-7 cells. b) In the presence of 100 μM intracellular cAMP, the Vm change displayed a non-voltage-dependent conductance in a doxycycline-treated IB3-8-3-7 cell, which was c) inhibited by the cystic fibrosis transmembrane conductance regulator inhibitor, glybenclamide (100 µM) when the cell was held at negative Vm. d) Current–voltage relationship in the absence (○) and presence (•) of glybenclamide.
Ibuprofen regulates CFTR function in a cAMP concentration-dependent manner
The effect of ibuprofen was studied on CFTR conductance with cAMP (100 µmol·L−1) in the ICS. Each cell was held at 0 mV and a voltage-ramp protocol was used to unveil the CFTR conductance (fig. 5⇓). Once a stable CFTR conductance was observed, adding ibuprofen (300 µmol·L−1) to the ECS produced a rapid inhibition of the conductance at Vm more hyperpolarised than -20 mV (figs 5a⇓ and b). Remarkably, ibuprofen also induced a robust and progressive enhancement of the CFTR current (fig. 5⇓), including a net increase of inward current at the cell's resting Vm (-30 mV; figs 5b–e⇓, and 6a).

a) In the presence of 100 μM intracellular cAMP, whole-cell cystic fibrosis transmembrane conductance regulator (CFTR) currents in a doxycycline-treated IB3-8-3-7 cell were revealed by a voltage-ramp protocol (in a sawtooth waveform between -100 and 100 mV). Immediately after the addition of 300 μM ibuprofen (▓) to the extracellular solution, it induced a blockade of CFTR current, revealed at negative membrane voltage (Vm). Besides the voltage-dependent blockade, ibuprofen gradually enhanced the CFTR current regardless of Vm. The dual effects of ibuprofen on the current–voltage relationship of CFTR at the time-points were as follows: b) ¶ versus #, c) + versus #, d) § versus #, and e) ƒ versus #.
The activity of CFTR can be upregulated or downregulated by a compound depending on the concentration of intracellular cAMP 15, and the concentrations of endogenous cAMP in epithelial cells are presumably at nM or low μM concetrations under physiological conditions 16. Alternatively, ibuprofen has a high affinity for plasma proteins and free ibuprofen composes <1% of the total plasma concentration 17. At clinical doses reaching 50–100 μg·mL−1 2, free ibuprofen in the plasma is only ∼1–2 μmol·L−1. The effects of ibuprofen at a low concentration (5 μmol·L−1) on CFTR activity were, therefore, tested in the presence of high (100 μmol·L−1) and low (5 μmol·L−1) concentrations of intracellular cAMP. At a high concentration of cAMP, ibuprofen caused a voltage dependent inhibition of the CFTR conductance (fig. 6b⇓). By contrast, at low cAMP concentrations, ibuprofen induced an enhancement of CFTR currents without any inhibition.
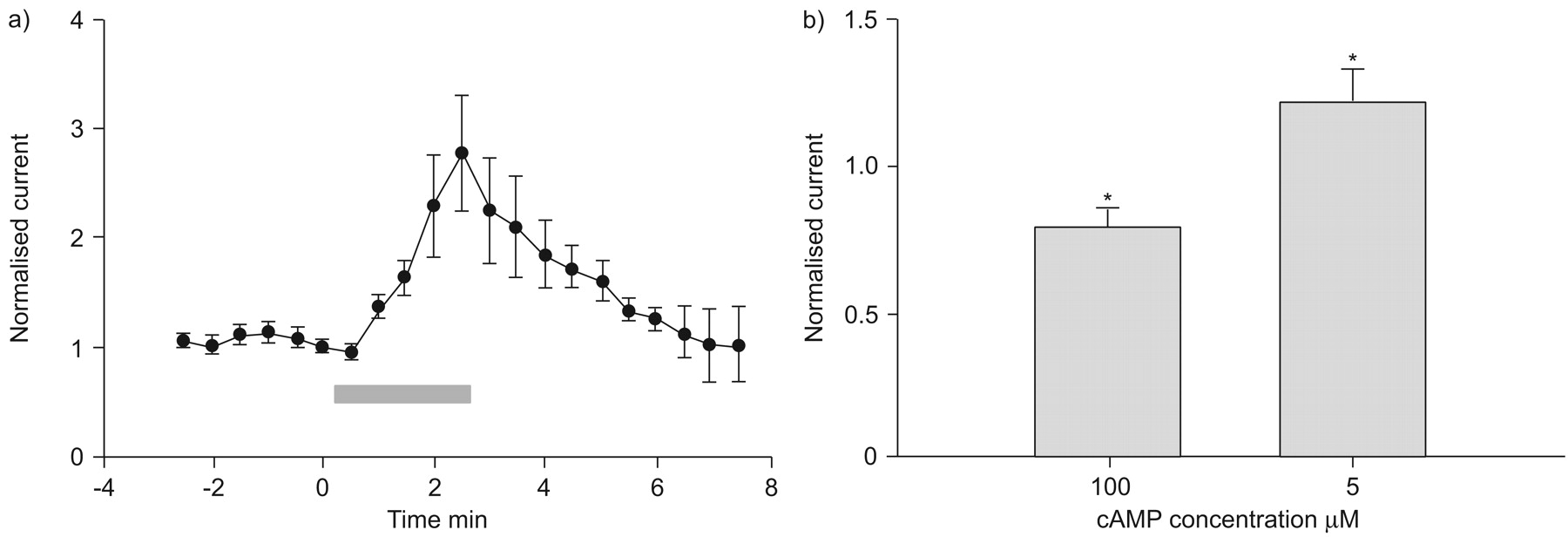
a) Cystic fibrosis transmembrane conductance regulator (CFTR) currents (at membrane voltage of -30 mV; n = 12) recorded before, during and after application of 300 μM ibuprofen (▓). The amplitude of current in the same cell at different time-points was normalised to that of the current just prior to ibuprofen application. b) Summary of the effect of 5 μM ibuprofen on normalised whole-cell CFTR current (measured at membrane voltage of −90 mV) in the presence of 100 μM (76±8.7% of control; n = 11) and 5 μM (148±15.6% of control; n = 5) intracellular cAMP. *: p<0.05 versus control.
The effect of ibuprofen on CFTR activity was then studied under perforated-patch recording conditions. The voltage ramp revealed a voltage-independent conductance in doxycycline-treated IB3-8-3-7 cells, which was enhanced by adding forskolin (5 μmol·L−1) to the ECS (fig. 7a⇓). In order to confirm that this forskolin-sensitive conductance in IB3-8-3-7 cells was mediated by Cl- flux, the cells were transfected with GABAAR subunits that form Cl- permeable channels 18. Application of GABA (30 μmol·L−1) evoked currents in the transfected cells when held at different values of Vm (fig. 7b⇓). Plotting GABA-induced currents against the holding Vm gave a linear current–voltage (I–V) relationship (fig. 7c⇓) and the calculated values for the reversal potential of GABA-currents (EGABA) was -23±2.2 mV (fig. 7d⇓). Alternatively, the reversal potential of the forskolin-sensitive conductance (Eforsk; fig. 7a⇓) was -22.4±2.3 mV (fig. 7d⇓), which was almost identical to EGABA. These data indicate that Cl- currents underlie the forskolin-sensitive linear conductance. Remarkably, ibuprofen (5 μmol·L−1) substantially enhanced this Cl- conductance (fig. 8a⇓ and b) without a blockade at hyperpolarised values of Vm (fig. 8a⇓). This ibuprofen-enhanced Cl- conductance was suppressed by glybenclamide (200 μmol·L−1; fig. 8a⇓) but persisted in the presence of 10 μmol·L−1 amiloride (ENaC blocker) and 10 μmol·L−1 bumetanide, a Na+–K+–2Cl--cotransporter inhibitor (n = 3 cells, data not shown), indicating that it is mediated by CFTR channels. Moreover, fast application of ibuprofen produced currents in the doxycycline-treated IB3-8-3-7 cell (see online supplementary figure 1a⇑) with a reversal potential at ∼-20 mV (see online supplementary figure 1b⇑). These combined data verify that ibuprofen enhances the tonic CFTR activity at endogenous concentration of cAMP.

Ibuprofen enhances cystic fibrosis transmembrane conductance regulator channel activity under physiological metabolic condition measured by perforated-patch recordings under voltage-clamp conditions with membrane voltage (Vm) maintained at 0 mV. a). Voltage ramp revealed a linear (non-voltage-gated) transmembrane conductance in a doxycycline-treated IB3-8-3-7 cell (#), which was enhanced by adding 5 µM forskolin to the extracellular solution (¶). b) Typical γ-aminobutyric acid (GABA)-evoked currents at different holding Vm (from -80 to 10 mV; for example +: -20 mV; and §: -30 mV) in a doxycycline-treated IB3-8-3-7 cell that was transfected with cDNAs encoding α1, β2 and γ2L subunits of GABAA receptors. c) Current–voltage relationship of GABA currents. d) Reversal Vm of GABA-current (EGABA; -23.4±2.2 mV; n = 6) and forskolin-sensitive currents (Eforsk; -22.4±2.3 mV; n = 5). ·····: 0. Data are presented as mean±sem.

Ibuprofen enhances cystic fibrosis transmembrane conductance regulator channel activity under physiological metabolic condition measured by perforated-patch recordings under voltage-clamp conditions with membrane voltage (Vm) maintained at 0 mV. a) Addition of 5 μM ibuprofen (▓) to the extracellular solution (ECS) enhanced the endogenous non-voltage-gated current at both negative and positive membrane voltage. The addition of 100 µM glybenclamide (▪) to ECS inhibited the ibuprofen-enhanced current. b) Normalised amplitude of voltage-ramp evoked currents (measured at -30 mV) before and during addition of ibuprofen (▓). Data are displayed as mean±sem. c) The enhancing effect in rat small intestinal epithelial cells on the voltage ramp-evoked current. d) Amplitude of current in the intestinal epithelial cells before (control; 260±26 pA; n = 5) and 3 min after (393±46 pA; n = 5) application of ibuprofen. Data are displayed as mean±sem. *: p<0.05.
Ibuprofen also enhanced CFTR activity in rat small-intestinal epithelial cells (figs 8c⇑ and d). The present authors conclude that ibuprofen enhances CFTR-channel function under physiological intracellular conditions. Moreover, meclofenamic acid, another NSAID, similarly enhanced the CFTR conductance of doxycycline-treated IB3-8-3-7 cells at depolarised Vm but inhibited the conductance at hyperpolarised Vm (see online supplementary figure 2⇑). The potentiation of CFTR function by NSAIDs does not seem to be directly associated with blockade of cyclo-oxygenase, an enzyme that converts arachidonic acid to prostaglandins, because the former inhibits CFTR activity 19.
Ibuprofen upregulates ▵F580-CFTR function
Whole-cell recordings were made on IB3-1 CF cells, in which few ΔF580CFTR proteins are targeted to the membrane surface. Indeed, in the presence of 100 μmol·L−1 cAMP in the ICS, sequential voltage ramps failed to reveal a CFTR-like conductance in these cells, even 10 min after whole-cell conformation (# in fig. 9a⇓).

Ibuprofen enhances ΔF508 cystic fibrosis transmembrane conductance regulator channel function in IB3-1 cells. a) Voltage ramp whole-cell recordings with 5 μM cAMP in the intracellular solution revealed a conductance in myoinositol-treated IB3-1cells but not in the untreated IB3-1cells (#: in the presence of 100µM cAMP). This voltage ramp-evoked conductance was enhanced by the addition of 300 µM ibuprofen (▓), and the ibuprofen-enhanced current could be suppressed by addition of 100 µM glybenclamide (▪). b) Under perforated-patch recording conditions, the voltage ramp revealed a nonlinear conductance in myoinositol-treated IB3-1 cells, which was enhanced by addition of 5 µM ibuprofen (▓) to the extracellular solution. c) Mean±sem endogenous membrane voltage measured under perforated-patch recording in the absence (-1.1±2.3 mV; n = 6) and presence (-32±4.8 mV; n = 5) of myoinositol in IB3-1 cells. Normalised amplitude of voltage ramp-evoked currents (measured at -30 mV) in myoinosital-treated IB3-1 cells before, during and after addition of d) 300 μM (n = 6) and e) 5 μM (n = 10) ibuprofen. *: p<0.05.
Treating epithelial cells with myoinositol, a constituent of phospholipids, can protect the ΔF508CFTR from degradation and enhances its surface expression 20. IB3-1 cells were treated with myoinositol for 18 h and voltage ramp revealed a CFTR-like conductance in myoinositol-treated cells (fig. 9a⇑, n = 9) when 5 μmol·L−1 cAMP was included in the ICS; this conductance was not evident when cAMP was omitted from the ICS. Also, it was blocked by glybenclamide (fig. 9a⇑). Given that ΔF508CFTR proteins are functional channels when targeted to the cytoplasmic membrane 21, the present authors conclude that this cAMP-induced glybenclamide-sensitive conductance in IB3-1 cells is mediated by the ΔF508CFTR; although, it exhibited a nonlinear I–V relationship at the hyperpolarised Vm 22. Importantly, this ΔF508CFTR-mediated conductance was enhanced by ibuprofen (figs 9a⇑ and d).
Perforated-patch recordings from IB3-1 cells treated with or without myoinositol were also made. The endogenous Vm of untreated IB3-1 cells was -1.1±2.3 mV whereas the myoinositol treatment increased Vm to -32±4.8 mV (fig. 9c⇑). Notably, voltage ramps revealed a conductance in the myoinositol-treated cells, which was increased by ibuprofen (5 μmol·L−1; fig. 9b⇑ and e). These results implied that ibuprofen enhances the function of ΔF508CFTR.
Ibuprofen upregulates CFTR activity through an intracellular mechanism
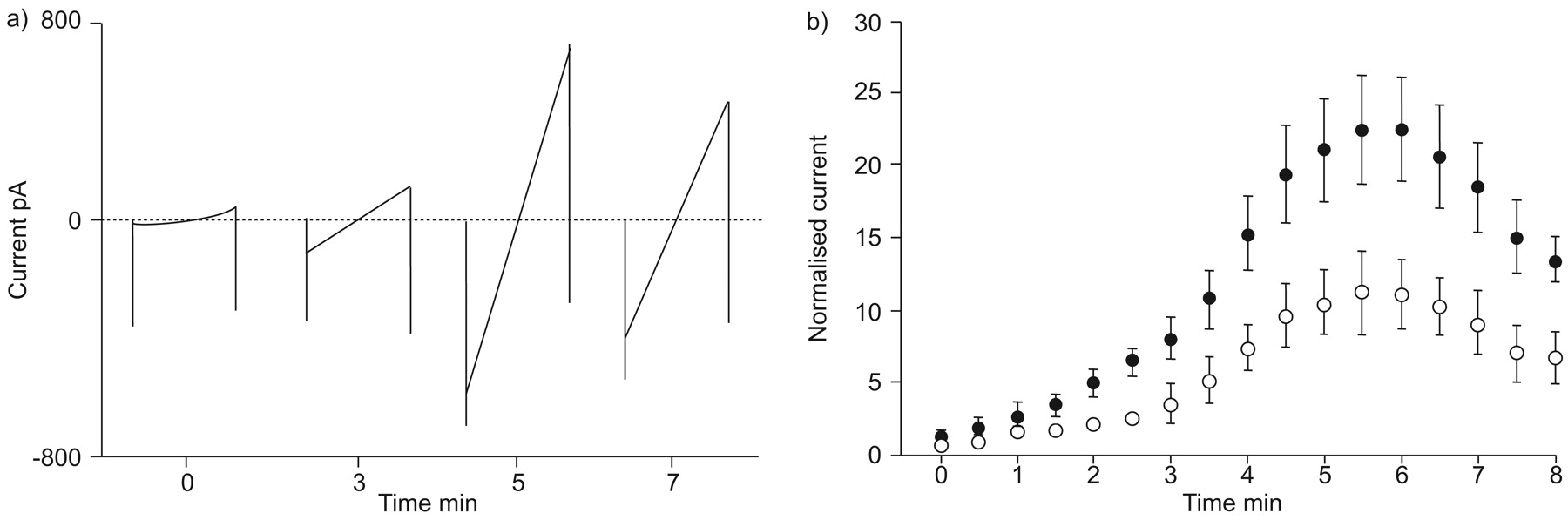
The enhancement of the CFTR conductance by ibuprofen developed slowly (fig. 5⇑) suggesting that the enhancing action might require access to the intracellular compartment. In order to test this idea, CFTR activity in doxycycline-treated IB3-8-3-7 cells was assessed sequentially using the voltage ramp protocol immediately after whole-cell compilation. Taking advantage of whole-cell configuration, ibuprofen was delivered (300 μmol·L−1, together with 100 μmol·L−1 cAMP) into the cell. A gradual increase in the amplitude of transmembrane conductance was observed, reaching the peak ∼4–6 min after whole-cell archetype, and then declined to a steady level ∼8–10 min after whole-cell model (fig. 10⇓). Notably, the I–V relationship of the increased currents was linear (fig. 10a⇓), indicating that intracellular ibuprofen does not produce a voltage-dependent blockade of CFTR. Moreover, plotting the normalised amplitude (measured at the Vm of -90 mV) of successively detected currents displayed a larger conductance in the presence of intracellular ibuprofen than in the absence of ibuprofen (fig. 10b⇓). These data suggest that ibuprofen upregulates CFTR activity via intracellular mechanisms. It was also found that in the presence of intracellular ibuprofen, addition of ibuprofen to the ECS produced a rapid inhibition of the current at negative values of Vm without enhancement at depolarised Vm (figs 11a⇓ and b). These data not only verify that ibuprofen enhances CFTR, by interacting with intracellular mechanism, but also indicate that its voltage-dependent blocking effect on CFTR is likely to be via extracellular access to the channel.

Ibuprofen exerts enhancing and inhibitory effects on cystic fibrosis transmembrane conductance regulator conductance by intracellular and extracellular mechanisms. a) Typical traces of voltage ramp-evoked currents recorded at different time-points of recording in the presence of 300 µM ibuprofen in the intracellular solution. b) Normalised amplitudes of voltage ramp-evoked currents at Vm of -90 mV of cells in the absence (○: n = 6 cells) and presence (•: n = 6 cells) of 300 µM ibuprofen in the intracellular solution. Currents were normalised to the one recorded immediately after whole-cell configuration.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Ibuprofen exerts enhancing and inhibitory effects on cystic fibrosis transmembrane conductance regulator (CFTR) conductance via intracellular and extracellular mechanisms. a) In the presence of 300 µM ibuprofen in the intracellular solution, 300 µM ibuprofen was added to the extracellular solution (▓) at 11 min after whole-cell configuration, and sequential traces illustrate the effect of extracellular ibuprofen on the evoked CFTR currents. b) Mean±sem amplitudes of the current recorded during perfusion of ibuprofen were normalised accordingly to the current evoked 30 s before addition of ibuprofen to the extracellular solution. The current amplitudes during perfusion of ibuprofen were 55±6.5, 84±3.0 and 97±2.6% of control size at -90, -60 and -30 mV of holding membrane voltage (Vm), respectively (n = 4). c) Successive traces show the effect of the addition of 300 µM ibuprofen to the extracellular solution (▓) on evoked CFTR currents in the presence of 10 µM genistein in the intracellular solution. d) Normalised mean±sem amplitude of the current recorded during perfusion of ibuprofen. The current amplitudes during perfusion of ibuprofen were 50±2.6, 81±3.4 and 98±2.5% of control size at -90, -60 and -30 mV of holding Vm, respectively (n = 4). **: p<0.001; ***: p<0.0001.
Genistein is a known CFTR-modulating reagent 23, 24, which enhances CFTR through binding to an intracellular domain 25 of the CFTR protein. With genistein (10 µmol·L−1) and cAMP (100 µmol·L−1) included in the ICS, adding ibuprofen (200 µmol·L−1) to the ECS, a voltage-dependent blockade, without any accompanying potentiation of the CFTR current, was observed (figs 11c⇑ and d). Using perforated-patch recordings it was further confirmed that application of genistein to the ECS enhanced the CFTR conductance and occluded the potentiation by ibuprofen (data not shown).
DISCUSSION
Results from the current study indicate that ibuprofen has at least two distinct effects on the CFTR conductance. First, at concentrations in excess of clinical doses, it can cause a rapid and voltage-dependent inhibition of CFTR activity, which is likely to be due to a direct channel blockade. Notably, ibuprofen causes voltage-dependent blockade of CFTR only when the channel is stimulated by a high concentration of cAMP. This is consistent with a previous study, which showed that high concentrations of ibuprofen inhibited the CFTR conductance, provided the channels were strongly activated by the forskolin or with the use of high concentrations of intracellular cAMP 3. Secondly, and most importantly, clinically relevant doses of ibuprofen substantially potentiate CFTR currents in the presence of low intracellular cAMP or in undisturbed intracellular conditions, which better reflect in vivo cell physiology. Taken together, the present findings have not only revealed potent and novel actions of ibuprofen in the upregulation of CFTR activity but also proved that NSAIDs regulate CFTR function in a channel activity-dependent manner through an intracellular interaction with this transmembrane protein.
The most common genetic defect in CF is the ΔF508 mutation 26. The present data show that ibuprofen can upregulate the activity of ΔF508CFTR provided that they are expressed on the cell surface. The present authors postulate that CF patients who have a reduced level of ΔF508CFTR in the epithelial cell surface would benefit from ibuprofen treatment. In addition, previous clinical studies on the determination of optimal dosages of ibuprofen for CF treatment were based on its effect on blocking neutrophil infiltration. The present results demonstrate that ibuprofen at clinical doses enhances CFTR channel activity in vitro. In order to assess its potential clinical benefits in CF patients, it will be necessary to confirm these findings using measurements of nasal potential difference.
Great efforts have been devoted to the search for drug candidates for cystic fibrosis treatment. For example, genistein is considered as a cystic fibrosis transmembrane conductance regulator-enhancing reagent 23–25 via direct interactions with cystic fibrosis transmembrane conductance regulator molecules. The ibuprofen-induced enhancement of the cystic fibrosis transmembrane conductance regulator can be occluded by genistein, which suggests that ibuprofen regulates cystic fibrosis transmembrane conductance regulator activity, possibly by interacting with the “genistein” binding site(s) on the cystic fibrosis transmembrane conductance regulator molecule. This notion needs to be demonstrated through further experiments. Genistein upregulates cystic fibrosis transmembrane conductance regulator function 23, 24. However, this compound is water-insoluble and strongly influences the endocrine and immune systems of newborn rats 27, thus rendering it inappropriate for clinical usage. In contrast, ibuprofen, a widely used and clinically approved nonsteroidal anti-inflammatory drugs, would be a much superior choice for treatment of cystic fibrosis patients.
Support statement
The present study was supported by Canadian Institutes of Health Research (CIHR) with operating grants (MOP-74653 and MOP-84517) to W-Y. Lu and by grants from CIHR and Canadian Cystic Fibrosis Foundation to J. Hu.
Statement of interest
None declared.
Acknowledgments
The authors thank B. Ye (Sunnybrook Health Sciences Centre, Toronto, ON, Canada) for assistance in cell culture. The authors would also like to thank M. Jackson (Dept of Physiology, University of Toronto, Toronto, ON, Canada) for comments on the manuscript.
Footnotes
-
This manuscript has supplementary data accessible from www.erj.ersjournals.com
- Received December 13, 2007.
- Accepted March 13, 2008.
- © ERS Journals Ltd
References










