





Abstract
Tobacco smoking continues to be the largest preventable cause of premature morbidity and mortality throughout the world, including chronic respiratory diseases such as asthma and chronic obstructive pulmonary disease. Although most smokers are highly motivated to quit and many smoking cessation therapies are available, cessation rates remain very low.
Recent research strongly suggests that variation in genetic background is an important determinant of smoking behaviour and addiction. Since these genetic variants might also influence the response to smoking cessation pharmacotherapies, it is likely that assessment of genetic background could be a promising tool to guide selection of the most effective cessation treatment for an individual smoker. Recently, it has been shown that genetic variants in the dopaminergic system, opioid receptors, the bupropion-metabolising enzyme CYP2B6 and the nicotine-metabolising enzyme CYP2A6 may play an important role in predicting smoking cessation responses to nicotine replacement therapy and bupropion treatment. Despite the progress that has been made, several challenges will still have to be overcome before genetically tailored smoking cessation therapy can be implemented in standard clinical practice.
Although the risk of cigarette smoking is well documented, tobacco smoking continues to be the largest preventable cause of disease and premature death throughout the world. It is estimated that there are currently still over 1.2 billion smokers worldwide, and this is expected to reach about 1.4–1.5 billion in 2010 and 1.6–1.9 billion by 2025 1–4. This is partly because of an increase in the adult population, and partly because of an increased consumption in low- and middle-income countries and among teenagers and females in high-income countries. Conversely, smoking prevalence among males in the high-income countries is declining 1, 4.
Inhalation of (cigarette) smoke has several deleterious effects on the airways, leading to and/or influencing chronic respiratory diseases such as asthma and chronic obstructive pulmonary disease (COPD). According to the latest World Health Organization estimates (in 2007), 300 million people have asthma and 210 million people have COPD. In contrast to other common smoking-related diseases, such as cardiovascular disease and cancer, chronic respiratory diseases are the only causes of death that are still increasing. By 2015, about 30% of the smoking-related deaths will probably be caused by chronic respiratory diseases 5.
Cessation reverses most adverse effects of smoking 6, 7. Smoking cessation as early as possible is important, but cessation at any age results in significant life extension 6, 7. Although the majority of smokers are highly motivated to quit, both smokers and healthcare practitioners are confronted with high relapse rates after initial successful smoking cessation attempts. Despite progress made in the (pharmacological) treatment of nicotine dependence, the efficacy of available treatments is limited. As shown in table 1⇓, only 15–30% of smokers continue to abstain from smoking 8–16. Therefore, multiple quit attempts are often required.
Efficacy of smoking cessation treatments
Recent research strongly suggests that smokers vary in their underlying genetic susceptibility to become addicted to smoking 17–23. Since pharmacogenetic therapies for smoking cessation are usually directed at the modulation of the pathways involved in smoking addiction, this genetic variation will probably also influence the efficacy of these smoking cessation therapies. Therefore, it seems that no medication will show efficacy for all smokers and the overall effectiveness of smoking cessation therapy might be increased if the therapy is targeted at those smokers most likely to respond. Research on the role of these inherited variations in the response to pharmacotherapy for nicotine addiction and smoking cessation may yield individualised treatments based on genotype. This is expected to result in a more efficient use of anti-smoking therapies, less frustration for smokers and more effort by healthcare providers in stimulating smoking cessation attempts. This will lead to increased cessation rates and, ultimately, in reduced deaths from chronic respiratory diseases caused by smoking.
The aim of the present review is to discuss the relevance of genetic profiling of genes involved in mediating smoking behaviour and addiction, as a tool to guide selection of an individualised and more efficacious smoking cessation therapy. In addition, the usefulness of this approach as a tool to prevent smoking-related chronic respiratory diseases will be discussed. The most important chronic respiratory diseases caused by smoking will be reviewed, along with the candidate genes contributing to the variation in smoking behaviour. Finally, the current knowledge on smoking cessation therapy will be discussed in the light of genetic background.
CHRONIC RESPIRATORY DISEASES ASSOCIATED WITH SMOKING
Smoking has been shown to be the main risk factor in the development of COPD and is known to influence the progression and treatment of both COPD and asthma. In the next section, a brief overview of the relationship of smoking with COPD and asthma is provided.
COPD
COPD is a leading cause of morbidity and mortality worldwide and results in an economic and social burden that is both substantial and increasing 24–26. Worldwide, COPD is the 12th most prevalent disease, the sixth most common cause of death and one of the few common causes of death increasing in incidence 24–26. COPD is predicted to rise to the fourth most prevalent disease and the fourth most common cause of death by 2035 5.
Smoking cessation is the most effective way to reduce the risk of developing COPD, since smoking accounts for 75–90% of COPD cases in the developed world, and exposure to tobacco smoke leads to a higher risk of developing COPD and a higher rate of disease progression 27, 28. The rate of mortality from COPD in nonsmokers is less than 10% of that of lifetime smokers 29, and the number of deaths from COPD increases depending on the number of cigarettes smoked 29. Even modest smoking (1–14 cigarettes·day−1) increases the rate of mortality from COPD by at least eight times compared with that of nonsmokers 29.
The impact of smoking cessation on respiratory symptoms, lung function, airway hyperresponsiveness and inflammation has been reviewed previously 30–33. It has been shown that when a smoker with declined lung function stops smoking, they will not regain lung function already lost, but smoking cessation is the only intervention proven to rapidly revert the rate of decline in forced expiratory volume in one second (FEV1) to the usual age-related decline, for both males and females at all stages of COPD. Smoking cessation improves long-term prognosis and also reduces respiratory symptoms such as cough and sputum production. Furthermore, airway reactivity seemed to improve in a 5-yr follow-up study, but several cross-sectional studies showed no reduction in airway reactivity to direct stimuli in ex-smokers compared with current smokers. Additionally, although some indirect markers of airway inflammation suggest a reduction in inflammation after smoking cessation, ex-smokers seem to have persistent airway wall inflammation and often remain symptomatic and experience frequent exacerbations of their disease 30–33.
Furthermore, a pooled meta-analysis has shown that steroid treatment is probably less effective in COPD patients who continue to smoke 34.
Asthma
The prevalence of asthma increased worldwide during the last quarter of the 20th century, particularly among children and adolescents, making it the most common chronic illness of childhood, with a prevalence varying from 0 to 30% in different geographical areas. In many countries a gradual increase in asthma mortality has been seen over the last 50 yrs 35–37.
The health effects associated with exposure to passive smoking have been widely studied and reviewed 38–45. Passive smoking, especially maternal smoking, in early childhood is reported to be associated with an increased risk for the development of asthma and more severe asthma symptoms, by impairing pulmonary function, enhancing airway reactivity and increasing pulmonary morbidity, leading to increased emergency department visits, hospitalisation rates and medication usage, and longer recovery periods after hospitalisation 38–45.
The health effects associated with active smoking have received less attention, since asthmatics are generally believed to be nonsmokers. However, the prevalence of smoking among adolescents with asthma has consistently been demonstrated to be equivalent to or even higher than rates among adolescents without asthma 42, 43, 46, 47, which means that about 15–35% of adult asthmatics are current smokers. Only a few reviews on the association of active smoking and asthma have been published 40–43, 47, 48. Smoking asthmatics have been shown to have worse symptom control than nonsmoking asthmatics, about four times more asthma attacks, an accelerated decline in lung function (FEV1) and increased pulmonary problems, such as a higher chance of respiratory failure and arrest, increased airway inflammation, more exacerbations, an exaggerated bronchoconstrictor response, a higher mortality rate after admission with a near-fatal asthma attack, and a higher chance of hospitalisation for intubation. The severity of these complications is positively related to the number of cigarettes smoked 40–43, 47, 48.
In addition, several studies have found that the efficacy of inhaled or oral corticosteroid treatment is also impaired in smokers with chronic asthma 47, 48.
CANDIDATE GENES FOR THE VARIATION IN SMOKING BEHAVIOUR
Tobacco smoking is believed to be a complex, multifactorial behaviour with both genetic and environmental determinants. While early reports suggested that the influence of heredity on smoking was modest, more recent studies have found significant genetic influences on several aspects of smoking behaviour. It has been demonstrated that genetic factors account for approximately 40–75% of the variation in smoking initiation, 70–80% of the variation in smoking maintenance, about 50% of the variance in cessation success and 30–50% of the variance in risk of withdrawal symptoms 17–23, 49, 50.
Variations in several genes have been suggested to contribute to smoking behaviour, and research has been focused on two broad classes of candidate genes: 1) genes that may influence the response to nicotine (e.g. nicotine metabolism, nicotinic receptors) and 2) genes that may predispose to addictive behaviour due to their effects on key neurotransmitter pathways (e.g. dopamine and serotonin) 23, 51–54.
In the next section, the most important candidate genes for the variation in smoking behaviour will be briefly discussed.
Genes influencing the response to nicotine
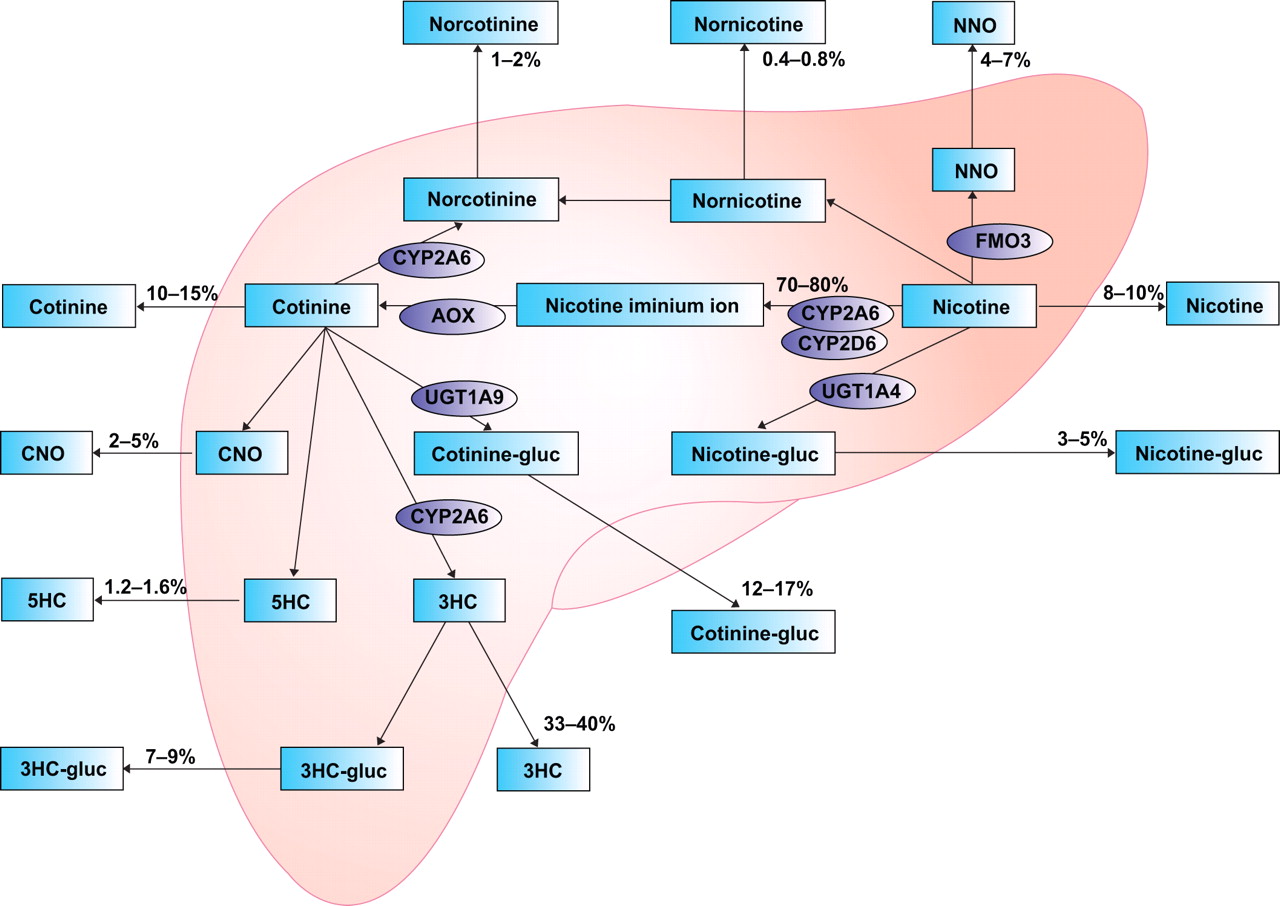
Nicotine is the primary reward component in tobacco products. Therefore, genes involved in the metabolism of nicotine are biologically plausible candidates for genetic studies of smoking behaviour, because they determine the levels and persistence of nicotine in the body. It is hypothesised that individuals with a high nicotine metabolism may experience fewer adverse reactions to their first encounter with nicotine and, therefore, may have a greater chance of continuing smoking and becoming addicted. Conversely, slow nicotine metabolisers may be less prone to initiate smoking because they may experience more adverse effects and would require fewer cigarettes to maintain nicotine titres at an optimal level once smoking is initiated 55. The major genes responsible for the metabolism of nicotine are the hepatic enzymes cytochrome P450 2A6 (CYP2A6) and cytochrome P450 2D6 (CYP2D6; fig. 1⇓).

Nicotine metabolism in the human liver. NNO: nicotine-1′-N-oxidase; CYP2A6: cytochrome P450 2A6; FMO3: flavin-containing monooxygenase 3; AOX: aldehyde-oxidase; CYP2D6: cytochrome P450 2D6; CNO: cotinine-N-oxide; UGT: uridine diphosphate-glucuronosyltransferase; cotinine-gluc: cotinine-glucuronide; nicotine-gluc: (S)-nicotine-glucuronide; 5HC: 5′-hydroxycotinine; 3HC: 3′-hydroxycotinine; 3HC-gluc: 3′-hydroxycotinine-glucuronide.
Of these, CYP2A6 is believed to be the most important predictor of the rate of nicotine metabolism, because it is responsible for roughly 90% of the metabolic inactivation of nicotine to cotinine 56, 57. A number of studies have shown that individuals carrying CYP2A6 variants that reduce the enzyme activity, determined in vivo via the measurement of the cotinine/nicotine or trans-3′-hydroxycotinine/cotinine ratios in blood or urine, are less tobacco dependent, smoke significantly fewer cigarettes per day and have an increased likelihood of quitting smoking 55, 58–62. Other studies have failed to detect these associations 63, 64 and a meta-analysis reviewing several studies on CYP2A6 genotype and smoking also found no association with smoking status and number of cigarettes smoked 65.
In addition, several studies have investigated the effects of CYP2D6 polymorphisms. Polymorphisms in CYP2D6 do not seem to be major determinants of nicotine metabolism in smokers except in ultrametabolisers (gene duplication) 66. This is probably because its catalytic activity towards nicotine is negligible in the presence of functional CYP2A6. Ultrametabolisers were found to be more likely to be heavy smokers 67. No relationship has been found for individuals with a poor CYP2D6 metabolism 67, 68, although one study did find a trend towards more poor metabolisers among males in the nonsmoking group 67, and another study reported that a poor CYP2D6 metabolism may reinforce smoking behaviour in committed smokers 69.
The pharmacological effects of nicotine are mediated by the activation of nicotinic acetylcholine receptors (nAChRs). High-affinity nicotinic receptors mainly contain the α4 (CHRNA4) and β2 (CHRNB2) subunits, and α4β2* (* indicates that another subunit may be included) is the most frequently encountered nicotinic receptor subtype. Several nAChR subunit genes have been examined for associations with smoking status (e.g. CHRNA4, CHRNA5, CHRNA7, CHRNB1, CHRNB2 and CHRNB3), but the functional relevance of the investigated variants in these genes is not yet known. Some evidence for an association with tobacco dependence and smoking status for variants in CHRNA5, CHRNA7, CHRNB1 and CHRNB3 70–72 has been provided. Evidence on the association of variants in the CHRNB2 and CHRNA4 genes is inconclusive 73–78.
Genes involved in the dopamine pathway
The mesolimbic dopamine system has been proven to play an important role in nicotine’s rewarding effects 79–82. Therefore, investigators have examined the association between smoking behaviour and variations in several genes involved in the dopamine pathway, such as dopamine receptors, the dopamine transporter and enzymes involved in dopamine synthesis and metabolism (fig. 2⇓).
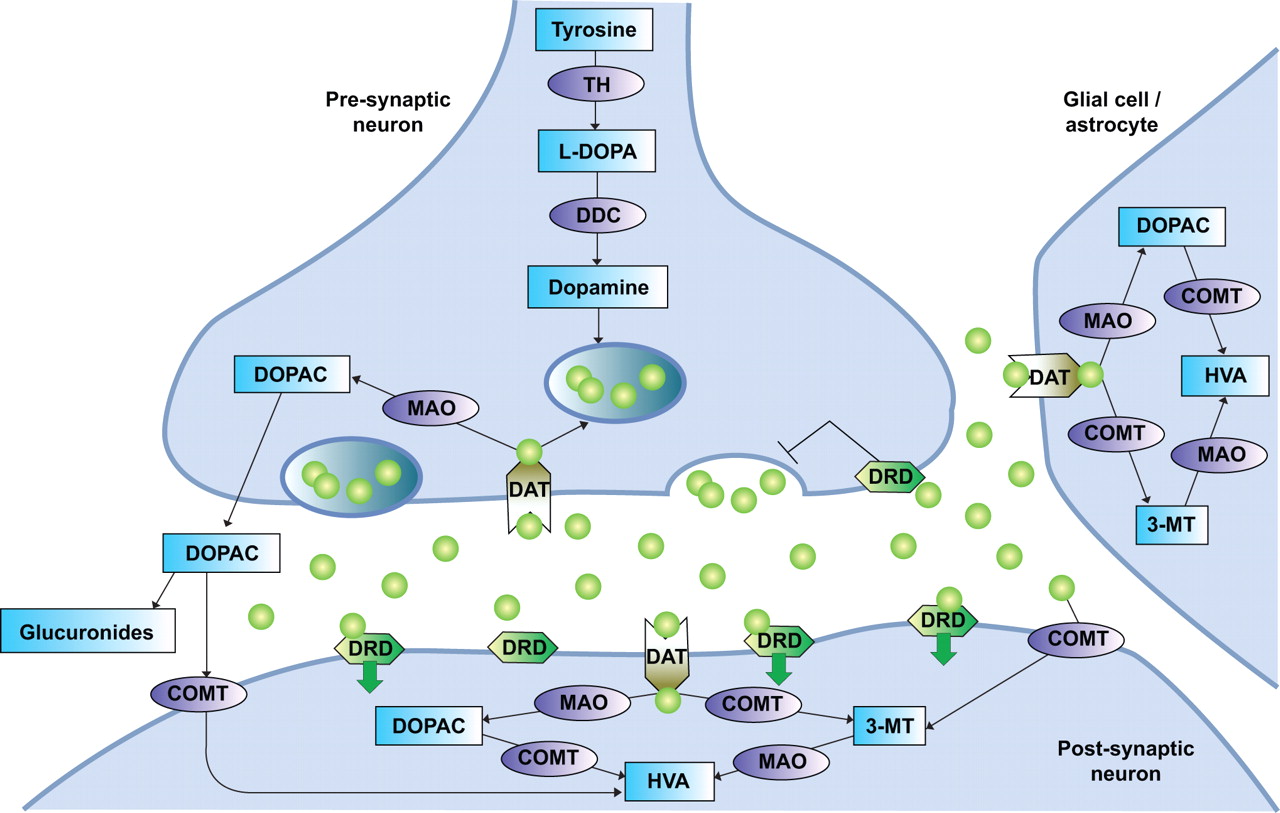
The dopamine pathway. TH: tyrosine hydroxylase; L-DOPA: L-3,4-di-hydroxy-phenylalanine; DDC: 3,4-dihydroxyphenylacetic acid (DOPAC) decarboxylase; MAO: monoamine oxidase; DAT: dopamine transporter; DRD: dopamine receptor; COMT: catechol-O-methyltransferase; HVA: homovanillic acid; 3-MT: 3-methoxytyramine. Green circles: dopamine.
Variants in several dopamine receptor genes (e.g. DRD1, DRD2, DRD4 and DRD5) have been detected and studied in relation to smoking behaviour. Overall, genotypes associated with reduced dopamine receptor expression or function seem to predict a higher chance of becoming a smoker, a younger age of onset, and fewer and less successful quit attempts 83–92. This is probably because subjects with reduced numbers of dopamine receptors may compensate for this deficiency by using nicotine to increase brain dopamine levels. However, the evidence concerning such results is still inconclusive 52, 93–95.
The dopamine transporter moves the dopamine released in the synapse into a neuron, glial cell or astrocyte to terminate the dopamine signal. A reduction in dopamine transporter levels, resulting in less clearance and greater bio-availability of dopamine, has been shown to be related to a lower chance of becoming a smoker, a lower nicotine intake and longer periods of smoking cessation 96, 97, but also, among African American smokers, to increased craving 98. Other studies failed to replicate these results 99, 100.
Several enzymes, such as tyrosine hydroxylase (TH), 3,4-dihydroxyphenylacetic acid decarboxylase (DDC), dopamine β-hydroxylase (DBH), catechol-O-methyl-transferase (COMT) and monoamine oxidase (MAO)-A and -B, are involved in the synthesis and metabolism of dopamine. Only limited data on the effects of variations in these enzymes on smoking behaviour are available. Associations between smoking and variations in genes for MAO-A, MAO-B, DBH and DDC have been found 101–107. No associations for most variants in TH have been reported; however, one variant seems to protect against smoking 101, 108–110. Contradicting results have been found for an increased activity COMT variant: some studies have found a positive association between the variant and nicotine dependence and smoking cessation 111–114, while others did not find an association 101, 112.
Genes involved in the serotonin pathway
The serotonin pathway is also under investigation in genetic studies of smoking, for several reasons. First, nicotine has been shown to increase the secretion of serotonin in the brain 115, 116. Secondly, increased serotonin levels have been associated with decreased food intake and weight gain, and have been shown to have an antidepressant effect 115. Furthermore, lower serotonin re-uptake has been associated with several behavioural traits (e.g. neuroticism, novelty seeking and anxiety-related personality traits) that are related to an increased incidence of smoking, increased nicotine dependence and difficulty in quitting smoking 117, 118. Candidate polymorphisms include those involved in serotonin biosynthesis (e.g. tryptophan hydroxylase (TPH)) and serotonin re-uptake (e.g. serotonin transporter (5-HTT)). Individuals homozygous for a variant of TPH, with an unknown effect, have been shown to be more prone to initiate smoking and to start smoking at an earlier age, but no effect on progression to nicotine dependence and smoking status has been found 119–121. Other studies have found that lower 5-HTT levels are associated with increased neuroticism in nicotine dependence 117, 118. Another study was unable to detect an association 122. Conversely, in a Japanese population, increased 5-HTT levels appeared to be associated with smoking 123.
INFLUENCE OF GENETIC VARIATIONS ON SMOKING CESSATION TREATMENT
Since the genetic background of mechanisms influencing smoking behaviour and addiction varies between smokers, it is a logical step to assess whether this genetic variation also determines the efficacy of smoking cessation treatment. Research on the role of such inherited genetic variation in the response to pharmacotherapy for smoking cessation opens avenues for individually tailored smoking cessation treatment based on genetic background. This might improve efficacy and minimise toxicity and side-effects of the treatments.
Most research on the role of genetic variation on smoking cessation pharmacotherapy has been directed to the two most widely accepted and licensed forms of smoking cessation therapy: nicotine replacement therapy (NRT) 111, 124–140 and the antidepressant bupropion (Zyban®) 129, 137, 141–149.
The relationship between genetic variation and efficacy of NRT can be illustrated by the following. First, it has been shown that smokers with genotypes associated with reduced dopamine levels (reduced dopamine receptor availability or function, high dopamine transporter levels and higher dopamine metabolism) achieve better quit rates with NRT compared with placebo 111, 124, 126, 127, 129, 134, 137, 139. Secondly, smokers who carry genetic polymorphisms associated with reduced nicotinic receptor (and possibly also dopaminergic) activity may experience greater benefit from nicotine spray (NS) compared with transdermal nicotine patches (TN), because of the greater rewarding effects of NS 133. Thirdly, smokers who have increased activity variants in the μ-opioid receptor (MOR) may have better success with the higher levels of nicotine delivered by TN compared with the lower levels of nicotine from NS 125, 136, 138, possibly only in combination with variants in MOR-interacting proteins 138. Fourthly, an increased nicotine metabolism (determined primarily by CYP2A6 genotype but probably not by CYP2B6 genotype) lowers quit rates with TN 128, 130, 135. Fifthly, variants in 5-HTT do not seem to influence the response to NRT 131, 132.
The efficacy of the antidepressant bupropion also seems to be related to a specific genetic background. First, genotypes associated with increased dopamine availability (increased or normal dopamine receptor availability or function, low dopamine transporter levels and low dopamine metabolism) predict a better response to bupropion 129, 137, 142–147, 149. Secondly, bupropion also seems to be more effective in smokers with a decreased bupropion metabolism (e.g. CYP2B6 decreased activity variants) 141, 148, but less effective in the presence of a decreased CYP2A6 metabolism 148. However, the influence of variants in the serotonin pathway on bupropion efficacy has not yet been investigated.
Despite all the progress that has been made in unravelling the pharmacogenetics of smoking cessation therapies, this research is still in its infancy and many challenges still have to be overcome before genetically tailored smoking cessation strategies can be effectively integrated into standard clinical practice.
First, so far, most studies have investigated only single genes. This approach will probably fail to fully determine the role of genetic variation in the individual susceptibility to smoking cessation therapies, since a large number of genes and polymorphisms in these genes are likely to contribute.
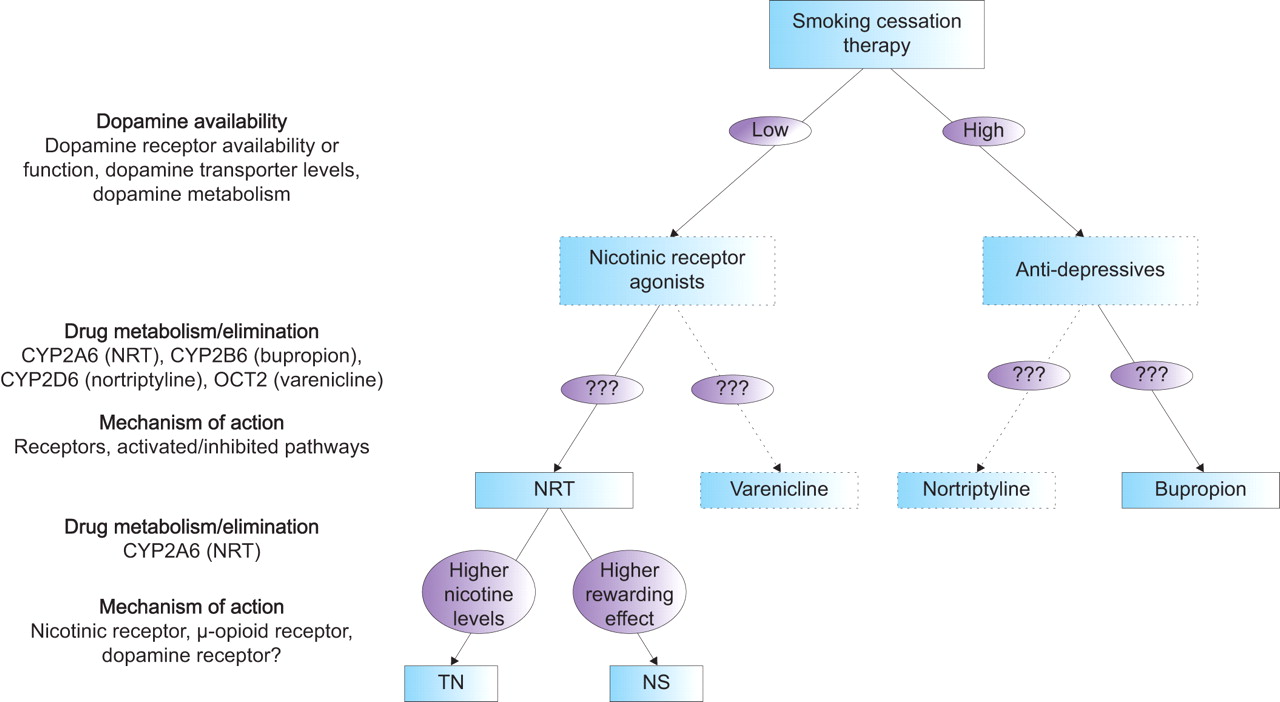
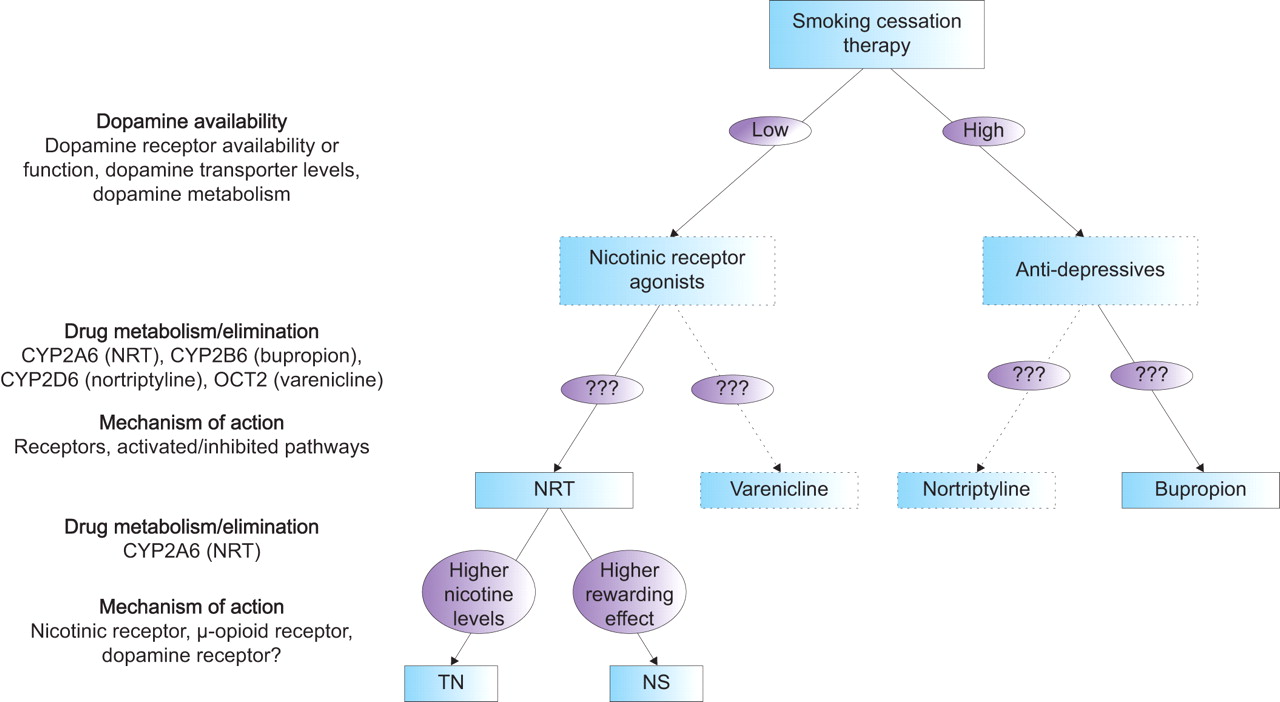
Secondly, until now, the pharmacogenetics have been investigated for only a couple of smoking cessation therapies. Newer compounds (e.g. varenicline), as well as current second-line medications for smoking cessation (e.g. nortriptyline), will also require investigation. Varenicline has been shown to bind with high affinity and selectivity at α4β2 receptors, thereby stimulating dopamine release while simultaneously preventing nicotine from binding (fig. 3⇓). Therefore, varenicline is expected to be more effective in smokers with genotypes associated with reduced dopamine availability, in a similar manner to NRT. Since nortriptyline has antidepressant properties, like bupropion, nortriptyline is expected to be more effective in smokers with genotypes associated with increased dopamine availability. However, differences in the metabolism or elimination of these drugs, as well as pathways involved in the mechanism of action, could make one drug more effective than the other or result in fewer side-effects in certain subgroups of smokers. In figure 4⇓ it is hypothesised how smoking cessation therapy might be genetically tailored based on present knowledge.

Mechanisms of smoking cessation treatments. Nicotine replacement therapy (NRT) partly replaces the nicotine (NIC) from smoking. Varenicline (V) blocks nicotinic acetylcholinic receptors (nAChRs), but still triggers dopamine release (green circles). Bupropion (B) inhibits re-uptake of dopamine, by occupying the dopamine transporter (DAT), thereby increasing the amount of dopamine to bind to post-synaptic dopamine receptors (DRDs). Nortriptyline (N) inhbits re-uptake of serotonin (red diamonds) by occupying the serotonin transporter (5-HTT), leading to increased amounts of serotonin to bind to post-synaptic serotonin receptors (5-HT-Rs).
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Hypothetical model for genetically tailored smoking cessation therapy. NRT: nicotine replacement therapy; CYP2A6: cytochrome P450 2A6; CYP2B6: cytochrome P450 2B6; CYP2D6: cytochrome P450 2D6; OCT2: organic cation transporter 2; TN: transdermal nicotine patch; NS: nicotine nasal spray.
Thirdly, genetic associations with tolerability and side-effects should also be examined. It is likely that some individuals are predisposed to have unusual reactions to drugs due to the presence of certain genetic defects. For example, it has been shown that variation in the DRD2 gene results in increased side-effects and thereby decreased adherence among females treated with bupropion 144. Certain subgroups of individuals may also exist who respond well to certain medications that are normally not well tolerated. For instance, individuals with a high nicotine metabolism may benefit from high-dose nicotine patches without experiencing the generally occurring side-effects.
Fourthly, a marked racial/ethnic diversity exists in smoking behaviour (e.g. age of initiation, smoking rate and level of dependence) and in the frequency of functional polymorphisms. To date, the vast majority of studies have been conducted with Caucasians, simply to avoid population stratification. Thus, the effect of genetic variants in other racial/ethnic groups should be investigated as well.
Fifthly, some research suggests that pharmacotherapies might work through different processes and/or be subject to different genetic influences in males and females. Therefore, the effect of genetic variations should be assessed for males and females separately.
Furthermore, the findings should be validated across independent trials, and prospective studies should be set up to fully confirm the effect of the variants.
Finally, several practical, policy and ethical considerations have to be addressed. Additional research should be conducted to examine the benefits, risks and challenges of conveying genetic information about smoking predisposition to the patient, clinicians and the public. Economic analyses of the cost-effectiveness of using genotype information to tailor smoking treatment would also be necessary and appropriate legal and regulatory frameworks should be set up to ensure privacy and to protect against genetic discrimination.
CONCLUSIONS
Health promotion and health education regarding tobacco use has made the public aware of the dangers of smoking and has resulted in increased cessation rates, but many smokers still continue to smoke, leading to high morbidity and mortality rates, especially from chronic respiratory diseases such as COPD and asthma. Although many of these smokers are highly motivated to quit, only a small proportion of individuals respond to the various treatments that are currently available to aid long-term smoking cessation. Since smoking behaviour has been shown to be influenced by genetic variations, it is expected that genetic variants might also influence smoking cessation success.
Based on recent research, it seems that genetic variants in several pathways related to smoking behaviour influence success rates of smoking cessation therapies. The effects of smoking cessation therapy might thus also differ considerably in subgroups carrying certain genetic variants. Therefore, a profile of genetic variants in these smoking-related pathways could possibly be used to predict in advance which smoking cessation therapy is likely to be most effective for an individual smoker. This could lead to a more effective use of smoking cessation therapies, resulting in fewer side-effects and increased cessation rates, and ultimately in reduced morbidity and mortality from chronic respiratory diseases such as COPD and asthma.
However, before genetically tailored smoking cessation therapy can be implemented in clinical practice, future studies should investigate the effect of multiple susceptibility genes, as well as their mutual interactions on several smoking cessation therapies, in large-scale, comparable trials in different ethnic/racial groups and different sexes. Additionally, prospective trials should be set up to fully confirm the effect of the variants.
Support statement
This work has partly been supported by a grant from the Netherlands Organisation for Health Research and Development (ZonMW, The Hague; Project No. 50-50101-96-404).
Statement of interest
None declared.
Footnotes
-
Earn CME accreditation by answering questions about this article. You will find these at the back of the printed copy of this issue or online at www.erj.ersjournals.com/current.shtml
- Received April 15, 2008.
- Accepted November 18, 2008.
- © ERS Journals Ltd
References










