





Abstract
Strategies to improve pulmonary endothelial barrier function are needed to reverse the devastating effects of vascular leak in acute respiratory distress syndrome. FTY720 is a pharmaceutical analogue of the potent barrier-enhancing phospholipid sphingosine 1-phosphate (S1P). FTY720 decreases vascular permeability by an incompletely characterised mechanism that differs from S1P. Here, we describe its barrier-promoting effects on intracellular signalling and junctional assembly formation in human pulmonary endothelium.
Permeability of cultured human pulmonary endothelial cells was assessed using transendothelial electrical resistance and dextran transwell assays. Junctional complex formation was assessed using membrane fractionation and immunofluorescence. Pharmacological inhibitors and small interfering (si)RNA were utilised to determine the effects of individual components on permeability.
Unlike S1P, FTY720 failed to induce membrane translocation of adherens junction or tight junction proteins. β-catenin, occludin, claudin-5 or zona occludens protein (ZO)-1/ZO-2 siRNAs did not alter FTY720-induced barrier enhancement. FTY720 induced focal adhesion kinase (FAK) phosphorylation and focal adhesion formation, with FAK siRNA partially attenuating the prolonged phase of barrier enhancement. Inhibition of Src, protein kinase (PK)A, PKG, PKC or protein phosphatase 2A failed to alter FTY720-induced barrier enhancement. FTY720 increased c-Abl tyrosine kinase activity and c-Abl siRNA attenuated peak barrier enhancement after FTY720.
FTY720 enhances endothelial barrier function by a novel pathway involving c-Abl signalling.
In the lung, endothelial cells (EC) serve as a semipermeable barrier between the vascular contents and pulmonary airspaces, and play a critical role in regulating tissue fluid homeostasis and the inflammatory response. Pulmonary endothelial permeability is primarily determined by a paracellular pathway that is regulated by a balance between intracellular contractile forces, and adhesive cell–cell and cell–matrix forces [1, 2]. A significant and sustained increase in vascular permeability is a hallmark of acute inflammatory processes, such as acute lung injury (ALI)/acute respiratory distress syndrome (ARDS), and is associated with significant mortality. Recent data indicate that there are >150,000 ALI cases per year in the USA, with a mortality rate of ∼35–40% [3]. Effective therapies for preserving or reconstituting the endothelial barrier are lacking despite their theoretic potential for ameliorating the vascular leak that characterises ALI.
Sphingosine 1-phosphate (S1P), a ubiquitous bioactive sphingolipid, has been identified as a robust barrier-enhancing agent [4]. Treatment of EC monolayers with S1P in vitro causes a rapid and sustained improvement in barrier function in a dose-dependent manner, as measured by transendothelial electrical resistance (TER). S1P infusion significantly attenuates lipopolysaccharide (LPS)-induced lung oedema and inflammation in murine and canine models of sepsis and ALI [5, 6]. Recent studies have identified transactivation of the S1P1 receptor by other barrier-enhancing agents as a common mechanism for improving endothelial barrier function [7]. Through activation of this Gi-protein coupled S1P1 receptor, S1P induces Rac-dependent peripheral translocation and colocalisation of cortactin and nonmuscle myosin light chain (MLC) kinase, MLC phosphorylation and cortical actin ring formation to produce improved barrier function [4, 8]. S1P also stimulates tyrosine phosphorylation of focal adhesion kinase (FAK) at a specific site (Y576) and subsequently causes focal adhesion (FA) formation and redistribution to the cell periphery, which probably contributes to improved barrier function [9, 10]. In addition, S1P may enhance barrier function by facilitating adherens junction (AJ) and tight junction (TJ) assembly [11–13].
FTY720, a structural analogue of sphingosine and S1P [14], is a promising treatment for multiple sclerosis that has been evaluated in recent phase III clinical trials [15, 16]. Like S1P, FTY720 significantly decreases LPS-induced pulmonary leak and inflammation in a mouse model of ALI [5]. We previously reported that FTY720 induced significant but delayed human lung endothelial barrier enhancement in vitro compared with the S1P response [17]. Unlike S1P, FTY720 did not induce MLC phosphorylation and subsequent cortical actin formation. Moreover, reduced expression of cytoskeletal effectors critical for S1P-induced TER elevation, Rac1 and cortactin, did not inhibit FTY720-induced TER elevation. In that study, reduction of S1P1 expression attenuated S1P- but not FTY720-induced TER elevations [17]. These results suggest a novel mechanism for FTY720-induced barrier enhancement that remains to be elucidated. There are two important reasons for studying, in detail, the effects of FTY720 on pulmonary EC barrier function. Unlike S1P, FTY720 has been evaluated in multiple clinical trials as a therapy for multiple sclerosis and transplant rejection, and soon may be widely available for clinical use. Thus, it has the potential for much more rapid translation into the intensive care unit as a possible therapy for ALI/ARDS than S1P. Secondly, improved understanding of the poorly characterised mechanism responsible for barrier enhancement by FTY720 may identify novel potential targets for the development of ALI therapies. In the present study, we further characterise the barrier-promoting effects of FTY720 on intracellular signalling and junctional assembly formation in lung endothelium and provide additional insights into barrier-regulatory pathways.
MATERIALS AND METHODS
Reagents
Unless otherwise specified, reagents were obtained from Sigma (St Louis, MO, USA). S1P (Biomol, Plymouth Meeting, PA, USA) was dissolved in 4 mg·mL−1 fatty acid-free bovine serum albumin (BSA). FTY720 was kindly provided by Novartis (Basel, Switzerland).
Antibodies
Anti-vascular endothelial (VE)-cadherin, anti-β-catenin, anti-zona occludens protein (ZO)-2, anti-pan FAK, anti-p120 and anti-paxillin antibodies were from Santa Cruz Biotechnology (Santa Cruz, CA, USA). Anti-occludin and anti-claudin-5 antibodies were from Zymed (San Francisco, CA, USA). The anti-vinculin antibody was from Sigma. The anti-phosphotyrosine 4G10 antibody was from Upstate Biotechnology (Lake Placid, NY, USA). Anti-phospho-FAK (Y397), anti-ZO-1 and anti-c-Abl (8E9) antibodies were from BD Pharmingen (San Diego, CA, USA). The anti-phospho-FAK (Y576) antibody was from Cell Signaling Technology (Danvers, MA, USA).
Pharmacological inhibitors
The pharmacological inhibitors of Src, protein kinase (PK)A, PKC, PKG and protein phosphatase (PP)2A used were PP2, dihydrochloride, 4-cyano-3-methylisoquinoline, Go6983, Ro-32-0432, Go6850, calphostin C and okadaic acid (EMD Chemicals, Gibbstown, NJ, USA).
Cell culture
Human pulmonary artery ECs (HPAECs) and human lung pulmonary microvascular ECs (HLMVECs) (Lonza, Walkersville, MD, USA) were cultured in Endothelial Basal Medium (EBM)-2 complete medium (Lonza) with 10% fetal bovine serum (FBS) at 37°C in a humidified incubator with 5% CO2, as previously described [4]. Passages 6–9 were used for experimentation.
Immunofluorescence
Confluent ECs grown on 35-mm glass-bottom petridishes (MatTek, Ashland, MA, USA) were incubated under various conditions as described for the individual experiments. ECs were then fixed in 4% paraformaldehyde for 20 min, permeabilised with 0.1% Triton X-100 for 2 min, washed in PBS, blocked with 2% BSA in PBS for 1 h and incubated with primary antibodies overnight at 4°C. After washing with PBS, ECs were incubated with donkey anti-mouse or donkey anti-rabbit immunoglobulin G antibody conjugated to AlexaFluor 488 or 568 (Invitrogen, Carlsbad, CA, USA) for 1 h at room temperature. Washed cells were then analysed using a Nikon Eclipse TE 300 microscope (Nikon Inc., Melville, NY, USA) and Sony Digital Photo camera DKC 5000 (Sony, Tokyo, Japan).
Small interfering RNA transfection
Negative control small interfering (si)RNA #2 (D-001810-02) and specific siRNAs (except claudin-5) were purchased from Dharmacon (Thermo Fisher Scientific, Lafayette, CO, USA; catalogue numbers L-003482-00 (β-catenin), L-011345-00 (occludin), L-007746-00 (ZO-1), L-009932-00 (ZO-2), L-003100-00 (c-Abl); siRNA sequence targeting FAK: sense GCGAUUAUAUGUUAGAGAUAGUU; antisense CUAUCUCUAACAUAUAAUCGCUU). Claudin-5 siRNA (sense: GGCUAAGAAUCUGCUUAGUtt; antisense: ACUAAGCAGAUUCUUAGCCtt) was purchased from Ambion (Austin, TX, USA). ECs (70% confluent) were transfected with 100 nM siRNA using DharmaFECT 1 transfection reagent (Thermo Fisher Scientific) according to the manufacturer’s protocol. Silencing efficacy assessment by Western blotting and subsequent experimentation were performed 72 h after siRNA transfection.
Transendothelial electrical resistance
ECs were trypsinised and seeded into polycarbonate wells containing evaporated gold microelectrodes, in EBM-2 with 2% FBS. After 24 h, cells had grown to confluence and TER measurements were performed using an electrical cell–substrate impedance sensing system (Applied Biophysics, Troy, NY, USA) as previously described [4]. TER values from each microelectrode were pooled at discrete time-points and plotted versus time as the mean±sem. Baseline TER values for HPAECs and HLMVECs used in these studies were 1,000–1,400 Ω prior to agonist stimulation.
Cell fractionation
Cell fractionation was performed as described previously [18]. ECs were solubilised in cytoskeleton stabilising buffer (CSK) (50 mM NaCl; 10 mM PIPES (piperazine-N,N′-bis(2-ethanesulfonic acid)), pH 6.8; 3 mM MgCl2; 0.5% Triton X-100; 300 mM sucrose; protease inhibitor cocktail I (Calbiochem, Gibbstown, NJ, USA)) for 20 min at 4°C. The cells were washed once with CSK buffer and scraped with a rubber policeman. The cell pellet was lysed in sodium dodecylsulfate (SDS) buffer (15 mM Tris, pH 7.5; 5 mM EDTA; 2.5 mM EGTA; 1% SDS) and incubated at 100°C for 10 min. After centrifugation at 14,000×g for 10 min, the supernatants were collected as Triton X-100-insoluble fraction and analysed by Western blotting.
In vitro c-Abl kinase assay
After treatment, 700 μg cell lysate, in radioimmunoprecipitation assay (RIPA) buffer, were immunoprecipitated with anti-c-Abl (8E9) antibody and protein G for 2 h. The beads were washed with lysis buffer once and with washing buffer (20 mM Tris-HCl, pH 7.2; 25 mM MgCl2; 5mM MnCl2; 0.4 mM EGTA) three times. 20 μL kinase reaction buffer (4 μL Src kinase reaction buffer (#20-121; Millipore, Billerica, MA, USA), 2.5 μL Crk–glutatione S-transferase (GST) (2 μg·μL−1; #14-468; Millipore); 1 μL adenosine triphosphate (1 mM); 12.5 μL H2O) were added to each tube. The reaction mixture was kept at 30°C for 20 min while shaking. Tyrosine phosphorylation of Crk–GST was detected by Western blotting with 4G10 antibody (Millipore).
Western blotting
ECs were solubilised in RIPA buffer with protease inhibitor cocktail I (for detecting phosphoproteins, phosphatase inhibitor cocktails II and III (Calbiochem) were added). Proteins were then separated in 4–15% SDS–polyacrylamide gels and transferred onto nitrocellulose membranes. After blocking for 1 h with 5% skimmed milk powder at room temperature, the blots were incubated with appropriate primary antibodies overnight at 4°C or 1 h at room temperature, followed by incubation with horseradish peroxidase-conjugated second antibodies for 1 h at room temperature. Visualisation of immunoreactive protein bands was achieved using enhanced chemiluminescence (Pierce, Rockford, IL, USA).
In vitro dextran permeability assay
We assessed fluorescein isothiocyanate (FITC)-labelled 40-kDa dextran (Sigma) permeability across EC monolayers plated on transwell inserts according to the manufacturer’s instructions (Chemicon, Temecula, CA, USA). Transwell inserts were coated with collagen for 1 h at room temperature and ECs were then seeded at a density of 1×105 cells·well−1 in a final volume of 400 μL EGM-2 with supplements. The inserts were placed into 24-well plates containing 500 μL medium and incubated overnight. To measure permeability, ECs were stimulated with agonists for 1 h, and 100 μL FITC–dextran was added into the insert and incubated for 2 h. The insert was then removed and 100 μL medium collected from the bottom chamber. The fluorescence intensity of samples was analysed on a Titertek Fluoroskan II Microplate Fluorometer (Diversified Equipment, Lorton, VA, USA) at excitation and emission wavelengths of 485 and 530 nm, respectively.
Statistics
The unpaired t-test was used to compare the means of data from two or more different experimental groups. Data are presented as mean±sem.
RESULTS
AJ and TJ proteins are not redistributed during barrier enhancement by FTY720
VE-cadherin is the major transmembrane protein of AJ in ECs and plays a critical role in maintaining and regulating endothelial permeability [19]. β-catenin and p120 catenin bind VE-cadherin and regulate AJ function [20]. FTY720 induces dose-dependent and sustained TER increases with a maximal effect at 1 μM at 1 h [17]. In order to investigate the effects of FTY720 on endothelial AJ protein distribution, confluent EC monolayers were stimulated with FTY720 and immunostained for VE-cadherin, β-catenin or p120 catenin. As shown in figure 1a, these AJ proteins are primarily localised at cell–cell boundaries. Unlike S1P [21], FTY720 failed to alter their distribution within 1 h, the timeframe in which maximal barrier enhancement occurs [17].

FTY720 does not induce adherens or tight junction protein rearrangement. a) Confluent human pulmonary artery endothelial cells (HPAECs) were stimulated with control vehicle (Con), or 1 μM FTY720 for 30 or 60 min, and immunostained with anti-vascular endothelial cadherin (VE-cad), β-catenin (β-cat) and anti-p120 antibodies. Images are representative of several independent experiments. b) Confluent HPAECs were stimulated with 1 μM sphingosine 1-phosphate (S1P) for 5 min or FTY720 for 5–60 min, and Triton-insoluble proteins were extracted as described. The amount of VE-cad and β-cat was detected by Western blotting. Each blot is representative of three independent experiments. c) Confluent HPAECs were stimulated with Con or 1 μM FTY720 for 60 min, or 1 μM S1P for 10 min, and immunostained with anti-occludin (Occ), anti-zona occludens protein (ZO)-1 and anti-ZO-2 antibodies. Images are representative of several independent experiments.
The anchorage of VE-cadherin, through catenins, to the actin cytoskeleton stabilises AJ and results in detergent resistance of AJ proteins. Therefore, ECs were stimulated with FTY720 and fractionated by Triton X-100 solution as described [18]. While S1P increased VE-cadherin and β-catenin in the insoluble fraction, FTY720 failed to alter the distributions of these AJ proteins (fig. 1b), suggesting that FTY720 does not increase the association of AJ proteins with the cytoskeleton. Thus, FTY720 induced neither cellular redistribution of VE-cadherin nor the anchorage of VE-cadherin to the cytoskeleton.
TJ complexes are another important class of cell–cell junctions that are responsible for regulating paracellular permeability and maintaining cell polarity [22]. TJs are composed of both transmembrane and intracellular molecules, including claudins, occludin, junctional adhesion molecules and ZO-1–3. Major transmembrane proteins in TJs include occludin and the claudin family of proteins, which consists of >20 members [23]. Of these, claudin-5 is expressed in large amounts in lung ECs [24]. FTY720 failed to alter the baseline distribution of occludin at 60 min, although S1P appeared to modestly increase peripheral localisation of occludin by 10 min (fig. 1c). Similarly, FTY720 treatment failed to alter the baseline distribution of claudin-5 at 60 min (data not shown). Furthermore, FTY720 failed to change the cellular distribution of ZO-1 and ZO-2 within 60 min, although S1P increased the membrane localisation of both proteins within 10 min (fig. 1c).
Decreased expression of AJ or TJ proteins fails to inhibit barrier enhancement by FTY720
β-catenin plays a pivotal role in stabilising and regulating the VE-cadherin complex [20, 25]. To further evaluate the role of AJ in FTY720-induced TER elevation, β-catenin expression was reduced by siRNA (online supplementary fig. 1). β-catenin siRNA decreased basal TER (data not shown) and significantly inhibited S1P-induced barrier enhancement (fig. 2b), but did not attenuate FTY720-induced TER elevation (fig. 2a). These data strongly suggest that AJ complex rearrangement is not essential for FTY720-induced TER elevation.
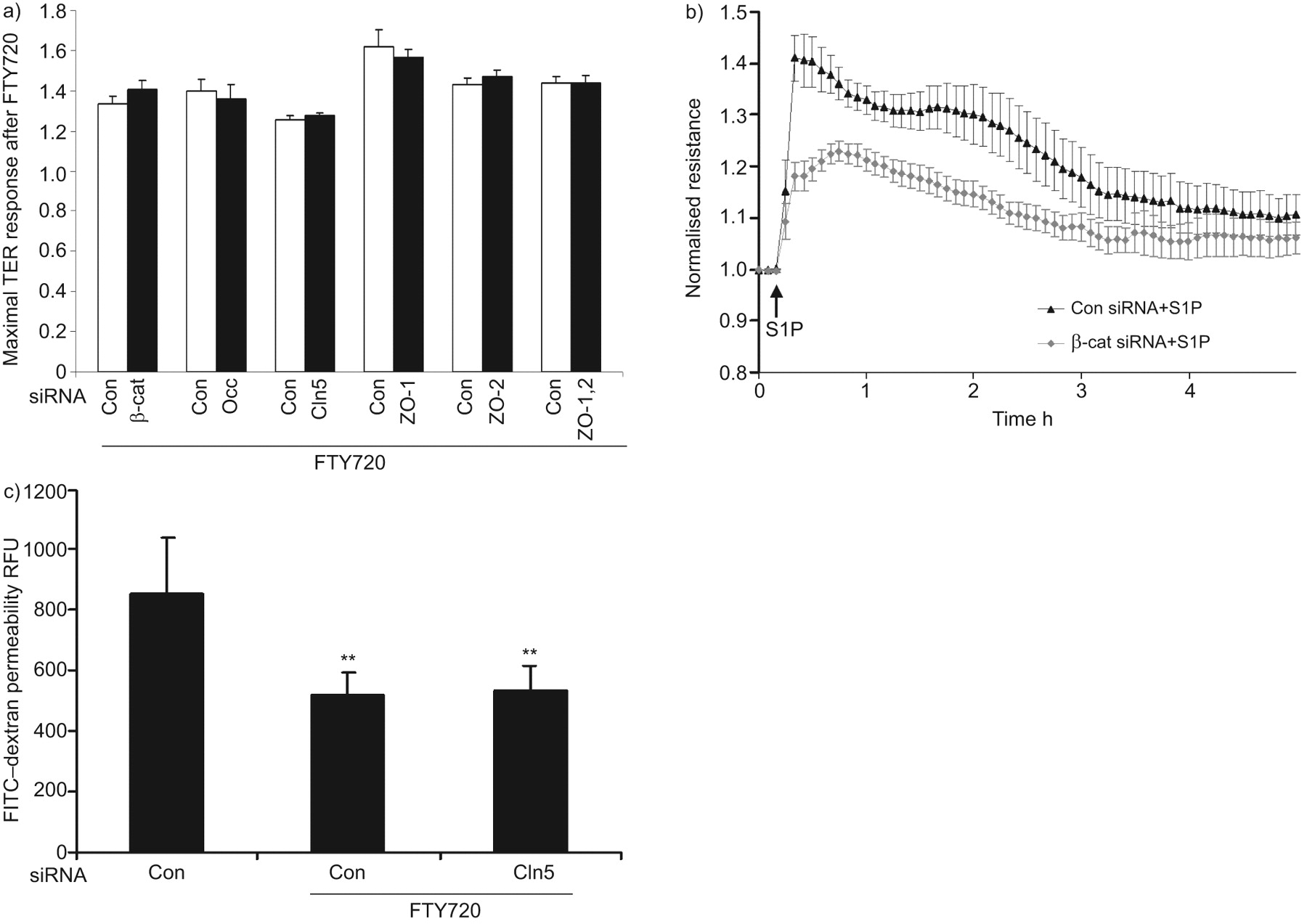
Barrier enhancement by FTY720 does not require β-catenin (β-cat), occludin (Occ), claudin-5 (Cln5), zona occludens protein (ZO)-1 or ZO-2. a) Human pulmonary artery endothelial cells (HPAECs) transfected with small interfering (si)RNA to β-cat, Occ, Cln5, ZO-1, ZO-2, both ZO-1 and ZO-2, or control siRNA (Con) were plated on gold microelectrodes and then stimulated with FTY720 (1 μM). Bar graphs represent the maximal transendothelial electrical resistance (TER) obtained after FTY720, relative to baseline resistance. Data are presented as mean±se. n = 3–5 per condition. There is no statistically significant difference between Con and targetting siRNAs. Western blots demonstrate representative downregulation by siRNA (online supplementary fig. 1). b) HPAECs transfected with β-cat siRNA were plated on gold microelectrodes and then stimulated with 1 μM sphingosine 1-phosphate (S1P). Data are presented as mean±sem. n = 3 per condition. c) HPAECs were transfected with 100 nM Cln5 or Con siRNA and seeded onto Transwell inserts. After FTY720 stimulation (1 μM), fluorescein isothiocyanate (FITC)–dextran was added into the top chamber and incubated for 2 h. The fluorescent intensity of the bottom chamber was analysed by fluorometry, as described in the Methods section. n = 3. RFU: relative fluorescence unit. **: p<0.01 versus control siRNA without FTY720. There was no statistically significant difference between Con and Cln5 siRNA when stimulating with FTY720.
In order to evaluate whether TJs participate in FTY720-induced TER elevation, expression of TJ proteins was downregulated by siRNA. Decreased occludin or claudin-5 expression (online supplementary fig. 1) did not affect either basal TER (data not shown) or FTY720-induced TER (fig. 2a). ZOs serve to link TJ with the actin cytoskeleton and play an important role in regulating TJ structure. However, neither ZO-1 nor ZO-2 siRNA altered basal TER (data not shown) or FTY720-induced TER elevation (fig. 2a). To exclude functional redundancy between ZO-1 and ZO-2 due to their structural similarity, we also simultaneously reduced the expression of these two proteins. The simultaneous knockdown of ZO-1 and ZO-2 failed to alter FTY720-induced TER elevation (fig. 2a). Furthermore, claudin-5 siRNA did not affect the ability of FTY720 to inhibit large molecule (dextran) permeability in a transwell assay (fig. 2c), which is consistent with the TER data. Thus, we conclude that the TJ proteins occludin, claudin-5, ZO-1 and ZO-2 are not essential for FTY720-induced barrier enhancement.
FTY720 induces FA rearrangement
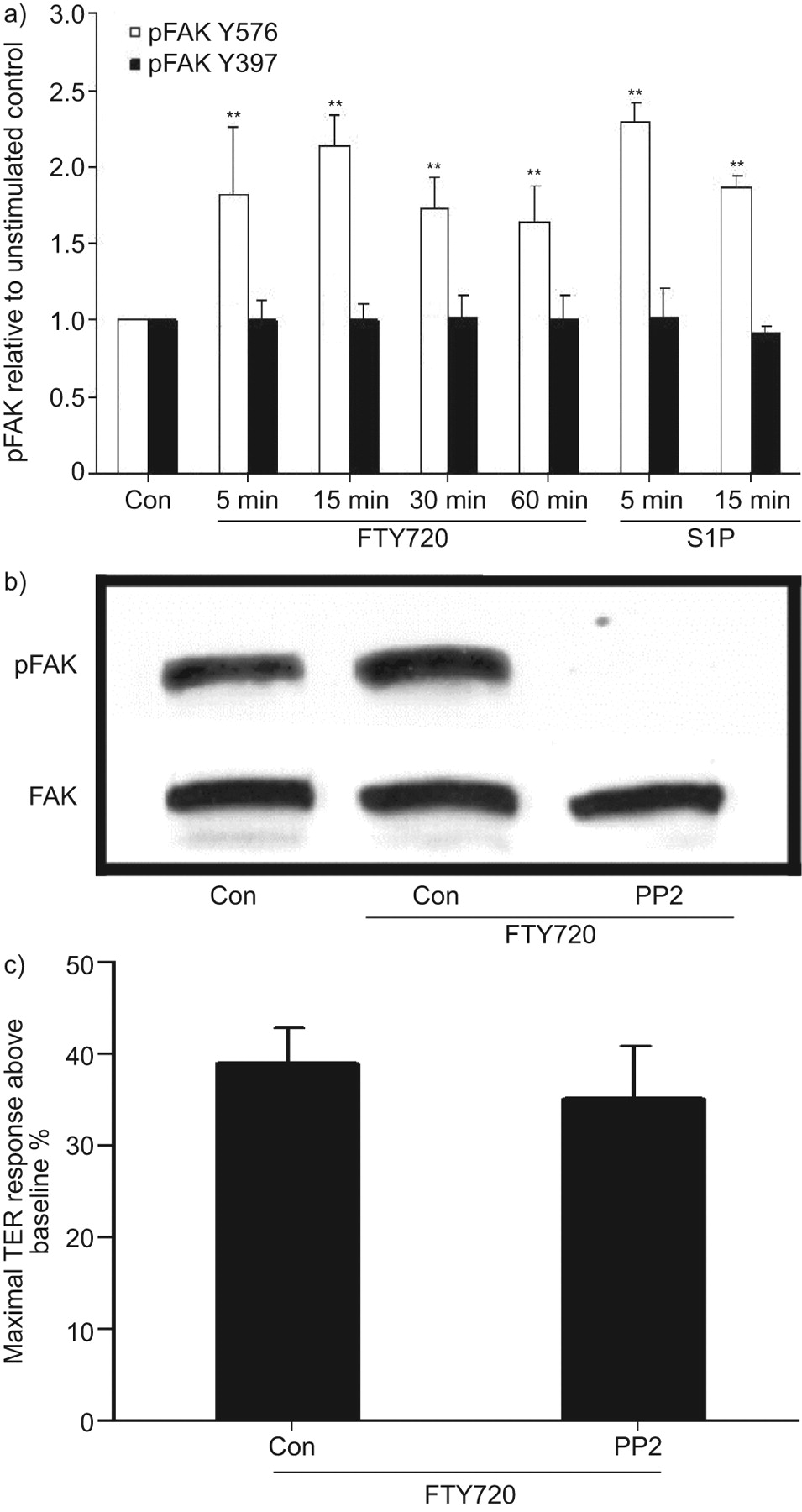
Recent evidence demonstrates that S1P induces significant FA rearrangement [9, 10]. In the present study, FTY720 appeared to increase the number of FA within 60 min (fig. 3a), as well as the amount of FA proteins paxillin and vinculin in the Triton X-100-insoluble fraction (fig. 3b), suggesting increased formation and stabilisation of FA. However, the functional importance of FTY720-induced FA rearrangement remains unclear, because neither paxillin nor vinculin siRNA attenuated TER increase by FTY720 (data not shown). The tyrosine kinase FAK plays a critical role in regulating FA structure and function 9]. FAK siRNA slightly reduced the basal TER (data not shown) but did not attenuate the initial rise in TER induced by FTY720 or its maximal elevation (fig. 3c). However, depletion of FAK significantly attenuated the sustained phase of TER elevation after FTY720 (i.e. after 4 h). It has been reported that phosphorylation of FAK at a specific site (Y576) plays an important role in the FA rearrangement that occurs during S1P-mediated barrier enhancement [9]. Thus, we next assessed whether FTY720 can induce similar phosphorylation of FAK. As shown in figure 4a, FTY720 quickly (within 5 min) induced significant phosphorylation of FAK at Y576, which persisted for ≥60 min (FTY720 did not increase phosphorylation of FAK at the thrombin-associated site, Y397). This phosphorylation was completely inhibited by pre-incubation with the Src inhibitor PP2 (fig. 4b). However, PP2 did not inhibit peak barrier enhancement by FTY720 (fig. 4c), which strongly suggests that Y576 phosphorylation of FAK is not required for the early and most potent effects of FTY720 on EC barrier function. Overall, these data suggest a possible role for FA in sustaining FTY720-induced TER elevation over time but no critical function during the initial increase in TER after FTY720.

FTY720 induces focal adhesion (FA) rearrangement. a) Confluent human pulmonary artery endothelial cells (HPAECs) were stimulated with 1 μM FTY720 for 60 min and then immunostained with anti-paxillin and anti-vinculin antibodies. Amplification of merged images suggests increased FAs after FTY720. Images are representative of several independent experiments. b) Confluent HPAECs were stimulated with 1 μM FTY720 for 30 or 60 min. The Triton-insoluble proteins were extracted as described. The amount of paxillin and vinculin was detected by Western blotting. The blot is representative of three independent experiments. c) HPAECs transfected with focal adhesion kinase (FAK) or control (Con) small interfering (si)RNA were plated on gold microelectrodes and then stimulated with FTY720 (1 μM). Data are presented as mean±sem. n = 3 per condition. Western blotting demonstrating representative downregulation of FAK by siRNA is shown in online supplementary figure 1.

Y576 phosphorylation of focal adhesion kinase (FAK) induced by FTY720 is not required for transendothelial electrical resistance (TER) elevation. a) Confluent human pulmonary artery endothelial cells (HPAECs) were stimulated with 1 μM FTY720 for 5, 15, 30 or 60 min, or 1 μM sphingosine 1-phosphate (S1P) for 5 or 15 min. Phosphorylation of FAK (Y576 or Y397) was detected using phosphorylation-specific antibodies. Data are presented as mean±se. n = 3 per condition. **: p<0.01 compared with control phosphorylation at Y576. There was no statistically significant difference for FAK phosphorylation at Y397. b) Confluent HPAECs were pretreated with 10 μM protein phosphatase (PP)2 for 60 min and stimulated with 1 μM FTY720 for 15 min. Phosphorylation of FAK (Y576) was detected using a phosphorylation-specific antibody. The blot is representative of three independent experiments. c) Confluent HPAECs plated on gold microelectrodes were pre-treated with 10 μM PP2 for 60 min and then stimulated with FTY720 (1 μM). Bar graphs represent the maximal TER obtained after FTY720 relative to baseline resistance. Data are presented as mean±se. n = 3 per condition. There was no statistically significant difference between control and PP2 pre-treated cells. Con: control.
Kinase/phosphatase signalling in FTY720-induced barrier enhancement: involvement of c-Abl tyrosine kinase
Previous investigations have demonstrated that intracellular signalling pathways involving PKA, PKC, PKG and PP2A regulate endothelial barrier function under various conditions [26–29]. Thus, we used multiple pharmacological inhibitors to inhibit these well-known intracellular signalling pathways: 4-cyano-3-methylisoquinoline (PKA inhibitor), H89 dihydrochloride (PKA and PKG inhibitor), Go6983, Ro-32-0432, Go 6850 and calphostin C (which combine to inhibit most PKC isoforms), and okadaic acid (PP2A inhibitor). None of these inhibitors significantly attenuated maximal FTY720-induced barrier enhancement (fig. 5).
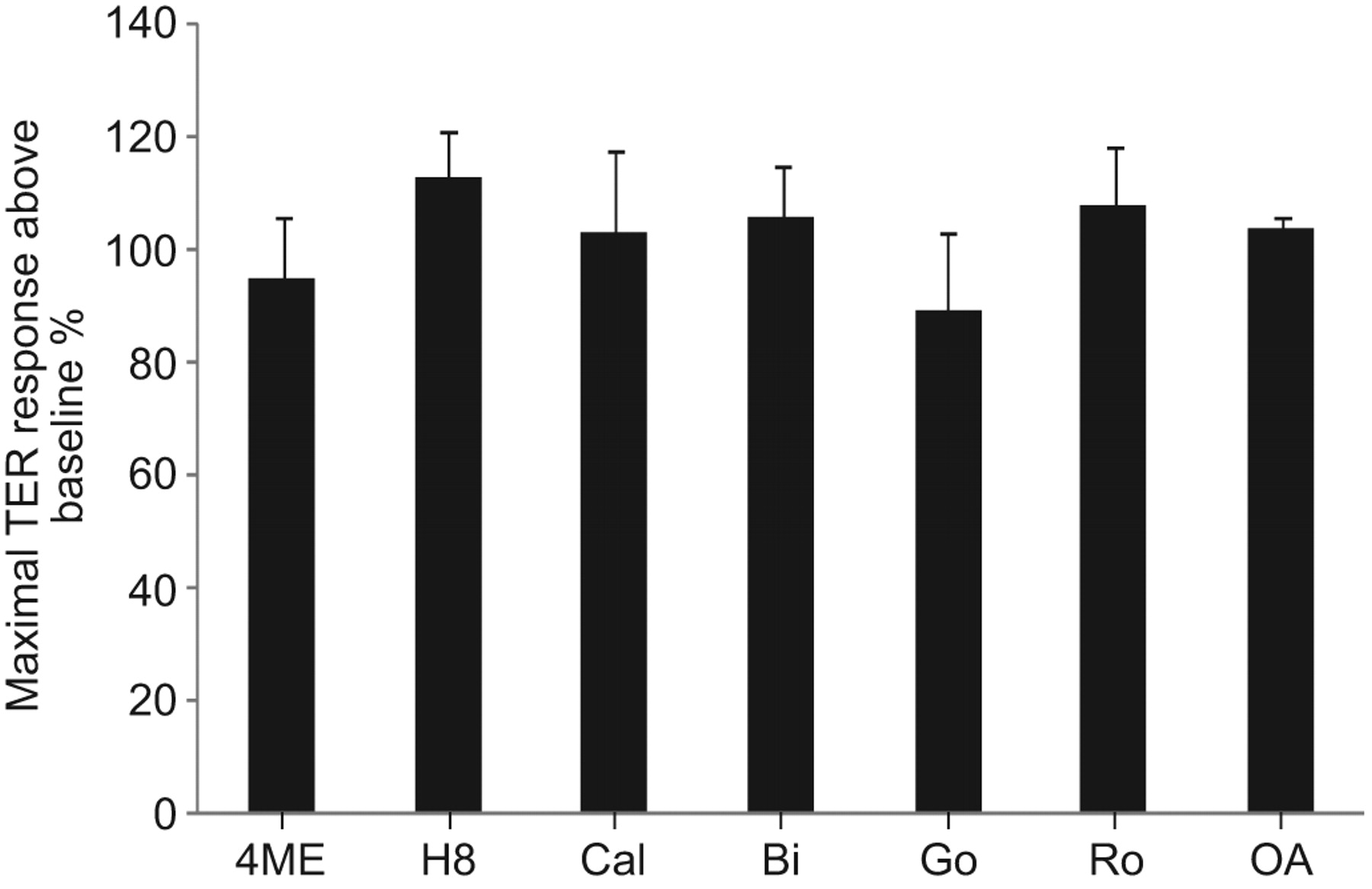
Barrier enhancement by FTY720 does not require protein kinase (PK)A, PKC, PKG or protein phosphatase (PP)2A intracellular signalling. Human pulmonary artery endothelial cells plated on gold microelectrodes were pre-treated with the following inhibitors or vehicle for 1 h: 4-cyano-3-methylisoquinoline (4ME) (25 μM; PKA inhibitor), H89 dihydrochloride (60 μM; PKA and PKG inhibitor), Go6983 (Go) (1 μM), Ro-32-0432 (Ro) (1 μM), Go 6850 (Bi) (1 μM) and calphostin C (Cal) (1 μM) (which combine to inhibit most PKC isoforms), and okadaic acid (OA) (2.5 nM; PP2A inhibitor). The range of inhibitor concentrations used was 10–100 times the median inhibitory concentrations provided by the manufacturers. Cells were then stimulated with FTY720 (1 μM). The maximal transendothelial electrical resistance (TER) increase of vehicle control plus FTY720 was set at 100%. n = 3 per condition. There were no statistically significant differences between any of the conditions shown.
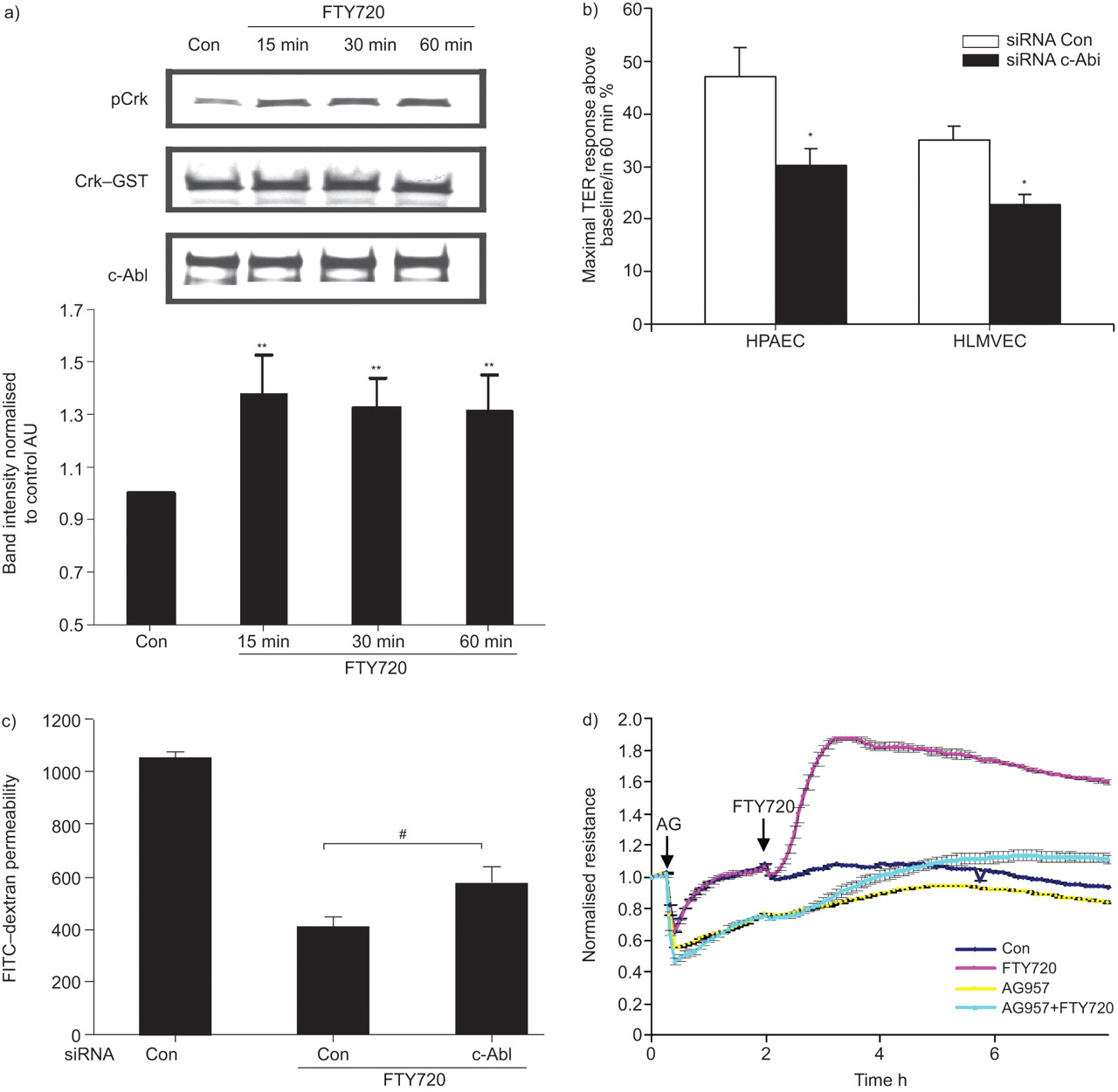
Our previously published data implicated tyrosine kinase signalling in FTY720-induced TER elevation, as the nonspecific tyrosine kinase inhibitor genistein significantly attenuated this response [17]. c-Abl is a ubiquitously expressed nonreceptor tyrosine kinase that is involved in the regulation of cell growth, survival and morphology [30]. FTY720 stimulation (1 μM for 15–60 min) of HPAECs significantly increased the capacity of immunoprecipitated c-Abl to phosphorylate recombinant Crk–GST (a known c-Abl target [31]), demonstrating that FTY720 increases c-Abl kinase activity during the timeframe within which it increases TER (fig. 6a). Importantly, c-Abl siRNA significantly attenuated FTY720-induced peak TER elevation by ∼40%, both in HPAECs and HLMVECs (fig. 6b) without changing the basal TER (data not shown). Well-described phenotypic differences have been described in the barrier regulatory responses of pulmonary macro- and microvascular endothelium to other barrier-altering agonists, such as thapsigargin [32] and thrombin [33], but our data demonstrate the similarity of the FTY720 response across these vascular beds within the lung. Furthermore, c-Abl siRNA significantly attenuated the ability of FTY720 to inhibit large molecule (dextran) permeability in a transwell assay (fig. 6c), confirming its importance in barrier regulation in a complementary approach to TER measurements. Finally, the pharmacological c-Abl inhibitor AG957 (20 μM) attenuated the FTY720 TER response (fig. 6d).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Barrier enhancement by FTY720 involves c-Abl tyrosine kinase. a) Human pulmonary artery endothelial cells (HPAECs) were stimulated with FTY720 (1 μM) or vehicle control (Con) for 15, 30 or 60 min. After treatment, c-Abl was immunoprecipitated with anti-c-Abl (8E9) antibody and incubated with Crk–glutathione S-transferase (GST) in kinase buffer as described in the Methods section. The tyrosine phosphorylation of Crk–GST (pCrk) was detected by Western blotting as a measure of c-Abl activity. The bar graph represents pooled densitometric results relative to vehicle control from three independent experiments. b) HPAECs or human lung pulmonary microvascular endothelial cells (HLMVECs) transfected with c-Abl siRNA or Con small interfering (si)RNA were plated on gold microelectrodes and then stimulated with FTY720 (1 μM). Bar graphs represent the maximal transendothelial electrical resistance (TER) obtained after FTY720 relative to baseline resistance. Data are presented as mean±se. n = 3 per condition. Western blotting demonstrating representative downregulation of c-Abl by siRNA is shown in online supplementary figure 1. c) HPAECs were transfected with 100 nM c-Abl or Con siRNA and seeded onto Transwell inserts. After FTY720 stimulation (1 μM), fluorescein isothiocyanate (FITC)–dextran was added into the top chamber and the cells were incubated for 2 h. The fluorescence intensity of the bottom chamber was analysed by fluorometry as described in the Methods section. n = 4. d) HPAECs were pre-incubated for 1 h with the c-Abl inhibitor AG957 (20 μM) or vehicle Con and then stimulated with FTY720 (1 μM) or vehicle Con. Data are representative of three independent experiments. *: p<0.05 versus Con siRNA; **: p<0.01 versus vehicle Con; #: p<0.031 versus Con siRNA.
These combined data strongly suggest that c-Abl expression and activity are necessary for maximal FTY720-induced barrier enhancement in human pulmonary ECs. As an initial exploration of the possible c-Abl downstream targets responsible for mediating barrier enhancement by FTY720, we downregulated expression of the well-described c-Abl target, Crk [31]. Although Crk siRNA greatly decreased basal TER, it did not inhibit FTY720-induced TER elevation (data not shown). Thus, the downstream targets of c-Abl responsible for mediating this FTY720 effect remain unclear. However, the well-known ability of c-Abl to regulate fibrous (F)-actin structures may be involved, because depolymerisation of F-actin with cytochalasin D partially inhibits TER elevation by FTY720 (online supplementary fig. 2).
DISCUSSION
FTY720 is currently in clinical trials as an immunomodulator for multiple sclerosis treatment and may soon be available as a therapeutic agent [15, 16]. Recently, we reported that FTY720 improved endothelial barrier function by a novel pathway different from that of S1P [17]. In the present study, we have further characterised the novel barrier-promoting effects of FTY720 on intracellular signalling and junctional assembly formation in human pulmonary ECs.
The present study has clearly demonstrated that the VE-cadherin complex does not participate in FTY720-induced barrier enhancement despite its well known role in EC barrier function. Although a recent study by Sarai et al. [34] reported increased peripheral localisation of both VE-cadherin and β-catenin in human microvascular ECs after prolonged exposure to FTY720 (3 h), in the present study, FTY720 did not cause redistribution of AJ proteins within the timeframe of maximal TER elevation (fig. 1a), nor did it induce tyrosine phosphorylation of VE-cadherin (data not shown) or increase the linkage between the VE-cadherin complex and the cytoskeleton (fig. 1b). Importantly, β-catenin siRNA did not attenuate FTY720-induced barrier enhancement even after 3 h of stimulation (fig. 2a). Interestingly, the direct functional role of VE-cadherin in mediating S1P-induced endothelial barrier enhancement also remains incompletely understood. In human umbilical vein ECs (HUVECs), S1P significantly increases the abundance of VE-cadherin and β-catenin at cell–cell contact regions [11]. However, recent data suggest that VE-cadherin is not necessary for the immediate TER increase (within minutes) after S1P, but may participate in the sustained phase that lasts for several hours [12].
Our data also suggest that TJ proteins are not essential for FTY720-induced barrier enhancement. TJs participate in endothelial barrier function to variable degrees along different segments of the vasculature [35]. For example, decreased expression of some claudins, the major transmembrane constituents of TJs [36], has been associated with decreased blood–brain barrier (BBB) function [37]. In particular, claudin-5 is specifically and highly expressed in ECs [24], and the BBB of claudin-5-deficient mice is selectively leaky for molecules <800 Da [37]. However, in the present study, claudin-5 siRNA did not change the basal TER of HPAECs or attenuate FTY720-induced barrier enhancement ([fig. 2a), nor did it alter permeability to larger molecules (dextran) after FTY720 in a transwell assay (fig. 2c). Downregulation of occludin, another transmembrane component of TJs [38], has been observed in BBB disruption and in vascular endothelial growth factor-treated retinal endothelial cells [39]. Our data indicate that HPAECs express occludin, but FTY720 did not induce its redistribution within these cells (fig. 1c). Furthermore, occludin siRNA neither changed the basal TER nor attenuated FTY720-induced barrier enhancement (fig. 2a).
Claudins and occludin are linked to numerous intracellular partners, including ZO-1, ZO-2, ZO-3, AF-6, protease-activated receptor-3, cingulin and 7H6 [40], which provide important connections to the underlying actin cytoskeleton. In HUVECs, S1P treatment caused ZO-1 redistribution to the lamellipodia and cell–cell junctions via the S1P1/Gi/Akt/Rac pathway, while the enhanced barrier function induced by S1P was attenuated by ZO-1 siRNA [13]. In the present study in HPAECs, we found that FTY720 failed to induce ZO-1 and ZO-2 redistribution (fig. 1c), and siRNA for ZO-1 and ZO-2 failed to attenuate FTY720-induced barrier enhancement (fig. 2a).
FAs are complex protein structures, consisting of integrins, actin-binding proteins and FAK [41], which contribute to EC barrier function by linking ECs to the underlying matrix [42]. Both depletion of FAK expression and expression of a kinase-deficient FAK mutant can impair EC barrier function [43]. Our data suggest that FTY720 increased the formation and stabilisation of FA (fig. 3). Furthermore, FAK siRNA inhibited the prolonged elevation in TER induced by FTY720 (>4 h) but did not alter peak TER occurring within the first hour. In order to further explore the possible role of FAK in FYT720-induced barrier enhancement, we assessed whether FTY720 induced phosphorylation of FAK at the specific site (Y576) known to mediate FA rearrangement that occurs during EC barrier enhancement by S1P [9]. Although FTY720 significantly induced phosphorylation of FAK at Y576 within the 1-h timeframe of maximal TER enhancement (fig. 4a), this phosphorylation can be completely inhibited by the Src inhibitor PP2 (fig. 4b) without affecting peak barrier enhancement by FTY720 (fig. 4c). These data strongly suggest that Y576 phosphorylation of FAK is not required for the early and most potent effects of FTY on EC barrier function. Overall, our data suggest that FA rearrangements and FAK may participate in the sustained phase of FTY720-induced barrier enhancement, but the mechanisms involved remain unclear and require further investigation.
PKC isoforms are important regulators of junctional permeability [26]. For example, PKCΔ has been reported to have a barrier-protective function by increasing FA contacts [44]. The activation of PKA promotes endothelial barrier integrity partially through its ability to stabilise EC cytoskeletal and adhesive structures [27], while PKG also has barrier-protective effects [45]. In the present study, we used multiple pharmacological inhibitors of PKA, PKG and PKC to screen for potential roles in the regulation of EC barrier function by FTY720. However, inhibitors for all of these pathways failed to attenuate FTY720-induced barrier enhancement (fig. 5). FTY720 is known to activate PP2A activity [46], another potential barrier-regulating protein [47]. However, inhibition of PP2A (via okadaic acid) had no effect on FTY720-induced TER elevation (fig. 5).
The nonreceptor tyrosine kinase c-Abl is ubiquitously expressed in mammalian cells and plays a critical role in the regulation of cell growth, survival and morphogenesis [30]. c-Abl can directly bind F-actin and interacts with a number of target effectors that regulate barrier-regulatory cytoskeletal structures, such as FA, lamellipodia, filopodia and membrane ruffles [30]. Abl/Abl-related gene knockout mouse embryos exhibit a significant defect in the actin latticework [48]. During cell adhesion, activation of c-Abl causes downregulation of Rap1–GTP, and cell rounding and detachment when Rho–ROCK1 is simultaneously activated [31]. The platelet-derived growth factor (PDGF)-induced dorsal-wave response requires c-Abl to phosphorylate the actin-binding protein cortactin on three critical tyrosine residues [49]. All of these observations strongly indicate that c-Abl is a critical mediator in regulating cytoskeletal organisation.
Current knowledge about the role of c-Abl in regulating EC barrier function is limited. One of the mechanisms by which FTY720 may have utility in treating leukaemia is through the dephosphorylation and inactivation of BCR–Abl fusion protein [50]. However, it is not clear how FTY720 regulates c-Abl in normal cells. The present study describes a novel role for c-Abl in mediating FTY720-induced pulmonary EC barrier enhancement. FTY720 significantly increased c-Abl kinase activity in cultured HPAECs (fig. 6a). Downregulation of c-Abl expression significantly inhibited FTY720-induced TER elevation in both HPAECs and HLMVECs (fig. 6b), as well as the ability of FTY720 to inhibit large molecule permeability in a transwell assay (fig. 6c). These data demonstrate an important role for c-Abl in achieving maximal barrier enhancement after FTY720, but it is not essential for all aspects of the FTY720 response because significant barrier enhancement still occurs when c-Abl expression is suppressed.
The mechanism by which c-Abl mediates this effect on EC barrier function is unclear. Multiple published studies have demonstrated the ability of c-Abl to regulate F-actin and cytoskeletal structures, including FAs, AJs, lamellipodia, membrane ruffles and filopodia (reviewed in [30, 51]). Because our study implicates c-Abl in EC barrier enhancement by FTY720 and EC barrier function is determined in large part by cytoskeletal structure, it is reasonable to hypothesise that c-Abl mediates the FTY720 effect via cytoskeletal rearrangement. For example, c-Abl has long been known to localise to FAs and phosphorylate various FA-associated proteins. However, our data indicate that FA proteins (FAK, vinculin and paxillin) are not essential for the early peak TER effect in which c-Abl participates (fig. 6). Similarly, inhibition of c-Abl results in disruption of cadherin-based AJs [52], but our data indicate that EC cadherin complexes are not involved in barrier enhancement by FTY720 in pulmonary EC (figs 1 and [2]). Therefore, it is unlikely that c-Abl contributes to EC barrier enhancement by FTY720 through regulation of FA or AJ junctional structures.
Another possible mechanism involves regulation of peripheral F-actin structures by c-Abl, which could theoretically mediate improved barrier function by FTY720. Multiple studies have demonstrated a role for c-Abl in modulating such structures. For example, cells deficient in Abl exhibit dramatically reduced membrane ruffling in response to PDGF, possibly because of decreased phosphorylation of the actin-binding protein cortactin [53]. Rapid peripheral cortical actin rearrangement and lamellipodial formation, mediated by Rac and cortactin, are critical aspects of EC barrier enhancement by S1P [8]. Moreover, c-Abl activity is known to regulate Rac and cortactin function [51], and our group has recently demonstrated a role for c-Abl in the S1P barrier-enhancing response in part via cortactin phosphorylation and cortical actin rearrangement [54]. In contrast, our previous work has demonstrated no role for Rac and cortactin in the FTY720 response, and immunofluorescence studies did not reveal peripheral actin rearrangement during EC barrier enhancement by FTY720 [17]. These observations suggest that dramatic peripheral actin rearrangement is not required for the FTY720 response, but the possibility remains that FTY720 may induce a modest amount of peripheral ruffling, mediated by c-Abl, which participates in barrier enhancement but is too subtle to be detected in confluent ECs by standard immunofluorescence imaging. The partial inhibition of FTY720-induced TER elevation by the actin-disrupting agent cytochalasin D is consistent with this hypothesis (online supplementary fig. 2).
To summarise, FTY720 enhances pulmonary EC barrier function by a novel pathway that does not require AJ or TJ protein complexes but does involve c-Abl signalling. Although the downstream targets of c-Abl responsible for this effect remain unclear and are the subject of ongoing investigation, these results suggest that modulation of c-Abl activity may represent a new therapeutic approach for regulating vascular permeability.
Acknowledgments
The authors thank L. Natarajan and S.M. Camp (both Institute for Personalised Respiratory Medicine, Section of Pulmonary, Critical Care, Sleep, and Allergy, University of Illinois at Chicago College of Medicine, Chicago, IL, USA) for superb technical support.
Footnotes
This article has supplementary material available from www.erj.ersjournals.com
Support Statement
This work was supported by grants from the National Heart Lung Blood Institute (NIH grants P01 HL 58064 (J.G.N. Garcia) and R01 HL 88144 (S.M. Dudek)).
Statement of Interest
None declared.
- Received March 25, 2010.
- Accepted October 1, 2010.
- ©ERS 2011
REFERENCES










