





Abstract
The aim of the current study was to define how cyclooxygenase (COX)-activity affects airway hyperresponsiveness (AHR) and inflammation using interventions with COX inhibitors at different time points during allergen challenge and/or prior to measurement of AHR in an eosinophil-driven allergic mouse model. Inflammatory cells were assessed in bronchioalveolar lavage (BAL) and AHR was evaluated as the total lung resistance to methacholine (MCh) challenge.
Administration of FR122047 (COX-1 inhibitor) during ovalbumin (OVA) challenge and prior to MCh challenge enhanced AHR without affecting the inflammatory cell response. In contrast, administration of lumiracoxib (COX-2 inhibitor) during the same time period had no effect on AHR but reduced the inflammatory cells in BAL. Nonselective COX inhibition with diclofenac both enhanced the AHR and reduced the inflammatory cells.
Administration of diclofenac only during OVA challenge reduced the cells in BAL without any changes in AHR, whereas administration of diclofenac only prior to MCh challenge enhanced AHR but did not affect the cells in BAL.
The present study implicates distinct roles of prostanoids generated along the COX-1 and COX-2 pathways and, furthermore, that inflammatory cells in BAL do not change in parallel with AHR. These findings support the fact that AHR and the inflammatory response are distinct and, at least in part, uncoupled events.
- Airway hyperresponsiveness
- allergic mouse model
- cyclooxygenase inhibition
- eosinophilic allergic reaction
- prostaglandins
The role of prostaglandins (PGs) and other cyclo-oxygenase (COX) products in asthmatic airway inflammation remains unclear. Despite a presumed pro-inflammatory action of most PGs in airways, studies in murine models of airway inflammation have shown that antigen-induced airway responsiveness in fact may be increased after pharmacological inhibition or gene deletion of COX iso-enzymes 1–4. There are two isoforms, COX-1 and COX-2, which catalyse the initial step in the formation of PGs and thromboxane (TXA) from arachidonic acid 5. Therefore, the first objective of the current study was to further define the role of the two iso-enzymes by intervention with selective COX-1 and COX-2 inhibitors in a mice model of allergic airway inflammation and airway hyperresponsiveness (AHR) to methacholine (MCh).
The models of airway inflammation generally involve three different phases. First, the animals are sensitised to produce antibodies towards a foreign antigen, most often ovalbumin (OVA). Thereafter, the animals are repeatedly challenged with inhalation of the same antigen to induce airway inflammation. This challenge induces an inflammatory process through an allergic reaction expressed as increased numbers of inflammatory cells in the lungs or bronchoalveolar lavage (BAL) fluid 6. The last part of the “mouse asthma” protocols involves assessment of the allergen-induced AHR by challenge with a bronchoconstrictor, most often MCh. It is often assumed that the increased inflammatory cell count in BAL is directly related to the appearance of AHR.
From previous studies reporting the effects of COX inhibition on airway inflammation and AHR in mouse models 1–4, it is not known at which time points in the sequence of events from sensitisation, induction of allergic inflammation and measurement of AHR that the COX acts. The secondary aim of the study was therefore to define the time points for effects of COX inhibition in this model by administration of the nonselective COX inhibitor diclofenac using three different strategies. Thus, its effects on AHR and BAL cell responses were compared when diclofenac was administered both during the OVA challenge and prior to the MCh challenge as well as when it was given either during the OVA challenge only, or only prior to the MCh challenge.
The airway responses were assessed by measuring AHR in anaesthetised mice based on the response of total lung resistance to MCh challenge and airway inflammation was assessed by measurement of inflammatory cells in BAL. To define mechanisms contributing to the changes in AHR and infiltration of cells into the airways, formation of key eicosanoids and cytokines in BAL were also investigated.
MATERIALS AND METHODS
Animals
Female BALB/c mice (8–13 weeks of age) were purchased from Charles River (Sulzfeld, Germany). The animals were housed in plastic cages with absorbent bedding material and were maintained on a 12 h daylight cycle. Food and water were provided ad libitum. All animal experiments were approved by the regional committee of animal experimentation ethics (Stockholm, Sweden).
Sensitisation and airway challenge
Mice were actively sensitised by i.p. injection of 10 μg OVA (grade II; Sigma-Aldrich, St Louis, MO, USA) in 1 mg Al(OH)3 (Sigma-Aldrich) in a total volume of 200 μL on days 0 and 7 (fig. 1⇓). Allergic airway inflammation was induced by challenge of 1% OVA (in PBS), or PBS (controls) aerosols on days 14, 15 and 16, delivered with an ultrasonic nebuliser (UltraNeb®; DeVilbiss, Somerset, PA, USA) for 30 min. Lung mechanics were assessed 24 h after the last OVA-challenge.

Allergic ovalbumin (OVA) mouse model: sensitisation and challenge protocol with the indication of drug administration. Drug interventions were administrated using three different strategies; 1) both during the OVA challenge and prior to methacholine (MCh) challenge; 2) during OVA challenge only; and 3) prior to MCh challenge only. BAL: bronchoalveolar lavage.
Intervention with COX inhibitors
Diclofenac sodium (1 mg·kg−1 body weight; nonselective COX inhibitor; Cayman Chemicals, Ann Arbor, MI, USA), FR122047 (5 mg·kg−1 body weight; selective COX-1 inhibitor; Cayman Chemicals), lumiracoxib (1 mg·kg−1 body weight; selective COX-2 inhibitor; SynphaBase, Muttenz, Switzerland) or solvent control was administered i.p. 1 h before each OVA challenge, 1 h before the MCh challenge and as an i.v. injection at the start of the anaesthesia (fig. 1⇑). Control mice received PBS-challenge instead of OVA aerosol. As a second control, either of the COX inhibitors was given in the same dose to mice sensitised to OVA but PBS-challenged. Since only the median inhibitory concentration (IC50) for diclofenac is described in mouse (IC50 COX-1/COX-2 0.5/0.35 µg·mL−1) 7 the dose used for FR122047 (IC50 COX-1/COX-2 0.028/65 µg·mL−1 in recombinant human assay) 8 and lumiracoxib (IC50 COX-1/COX-2 67/0.13 µg·mL−1 in human whole blood assay) 9 were selected from in vitro studies in mice and rats 9 in which they were both effective and selective.
To study the time-point of the effect of COX-inhibition, diclofenac was also administered in the same dose (1 mg·kg−1 body weight) either only during the OVA challenge (intranasal 100 µg OVA) or acute prior to the MCh challenge and as an i.v. injection at the start of anaesthesia without administration during the OVA challenge.
Determination of AHR
Mice were anaesthetised with pentobarbital sodium i.p. (90 mg·kg−1 body weight; from Apoteket Produktion and Laboratorier AB, Stockholm, Sweden), tracheostomised with a metal 18-gauge cannula and placed on a 37°C heating pad to maintain body temperature during the anaesthesia. Mice were mechanically ventilated in a quasi-sinusoidal fashion 10 with an animal ventilator (FlexiVent®; Scireq, Montreal, QC, Canada) 11 at a frequency of 2.5 Hz and a tidal volume of 12 mL·kg−1 body weight. In this mode, the pressure waveform is only sinusoidal during inflation not deflation, mimicking conventional rodent ventilators. Once the ventilation began, bilateral thoracotomies were performed to equalise the pleural pressure to atmospheric pressure and to exclude any chest wall contribution to pulmonary mechanics. The positive end-expiratory pressure was set to 3 cmH2O. To stabilise the baseline respiratory lung resistance (RL) and ensure similar volume history, four sigh manoeuvres to three times the tidal volume were performed at the beginning of the experiment defined as incremental increase and decrease of lung volume during 16 s. After 5 min resting, the experiment was started and increasing doses (0.03, 0.1, 0.3, 1 and 3 mg·kg−1 body weight) of MCh (acetyl-β-methylcholine chloride; Sigma-Aldrich) was injected through the tail vein. RL and pulmonary compliance (CL) were measured by assuming a single-compartment linear model and multiple linear regressions at a sinusoidal frequency of 2.5 Hz every eight breaths for 3 min after each injection 12. Changes in reactivity and sensitivity were assessed using nonlinear regression analysis to calculate the maximum responses (Emax) and effective dose for half maximal response (ED50). Since a dose of 10 mg·kg−1 body weight of MCh caused such bradycardia that it resulted into cardiac arrest with no further increase in AHR, the Emax was reached at 3 mg·kg−1 or, in some experiments, even earlier because of the same phenomenon. CL is expressed as the maximal decrease to each MCh dose.
BAL
BAL was performed in all controls and treated animals directly after AHR measurements. In short, a total volume of 1 mL ice cold PBS containing 0.6 mM EDTA was used to lavage the lungs three times. Red blood cells were lysed by resuspending the cells in 100 µL lysis buffer (150 mM NH4Cl, 10 mM KHCO3 and 0.1 mM EDTA at pH 7.2) for 2 min at room temperature followed by washing in 1 mL PBS. The total number of cells was then counted and calculated back to cells·mL−1 BAL. For differential cell counts, a minimum of 300 cells were counted per BAL sample.
Measurements of released eicosanoids
PGD2 (PGD2-MOX; Cayman Chemicals, Ann Arbor, MI, USA), PGE2, TXA2 and cysteinyl leukotrienes (CysLTs) were measured in BAL fluid using enzyme immuno assay (EIA; Cayman Chemicals). All samples were assayed in duplicate. EIA TXA2 was measured as the stable metabolite TXB2. CysLT were measured as leukotriene (LT) E4, the end metabolite of LTC4 and LTD4. The assay detection limits for the different mediators were 3.9 pg·mL−1 for PGD2 and 7.8 pg·mL−1 for PGE2, TXB2 and LTE4. Results below detection limits were set as detection limit in the statistical evaluation.
Measurement of released cytokines
The concentrations of interleukin (IL)-4, IL-5, IL-10, IL-13, tumour necrosis factor (TNF) and interferon (IFN)-γ in BAL fluid were analysed in all control and treated animals measuring fluorescence-labelled beads (Cytometric Bead Array; BD Biosciences, San Diego, CA, USA) by flow cytometry, following the manufacturer's protocol, and compared with known standards. The detection limit was 5 pg·mL−1.
Immunoglobulin E and immunoglobulin G1 analysis
Blood was collected by cardiac puncture for measurement of antibody titres using ELISA and plates coated with 5 µg·mL−1 OVA (grade II, Sigma-Aldrich) as described previously 13. As no OVA-specific immunoglobulin (Ig) E and IgG1 standards exist, values were expressed in units of optical density (OD).
Statistical analysis
All data are presented as mean±sem. Differences among the treatment groups were assessed by one-way or two-way ANOVA. Significant ANOVAs were further analysed by Bonferroni post hoc test. Two-way ANOVA was used to analyse the dose–response curves. Otherwise one-way ANOVA was used in all other statistic analysis. ED50 was analysed as log-values to follow normal distribution. A p-value of <0.05 was considered significant. Statistical analysis and graphs were performed in Graph Pad Prism (version 5.0; GraphPad Software Inc., San Diego, CA, USA).
RESULTS
Sensitisation
Sensitisation was confirmed by increased levels of OVA-specific IgE (OD 0.17±0.02; p<0.01) and IgG1 (OD 0.69±0.08; p<0.001) compared with naïve mice (OD 0.07±0.01 and 0.05±0.00, respectively).
Intervention with COX inhibitors during both OVA and MCh challenge
AHR
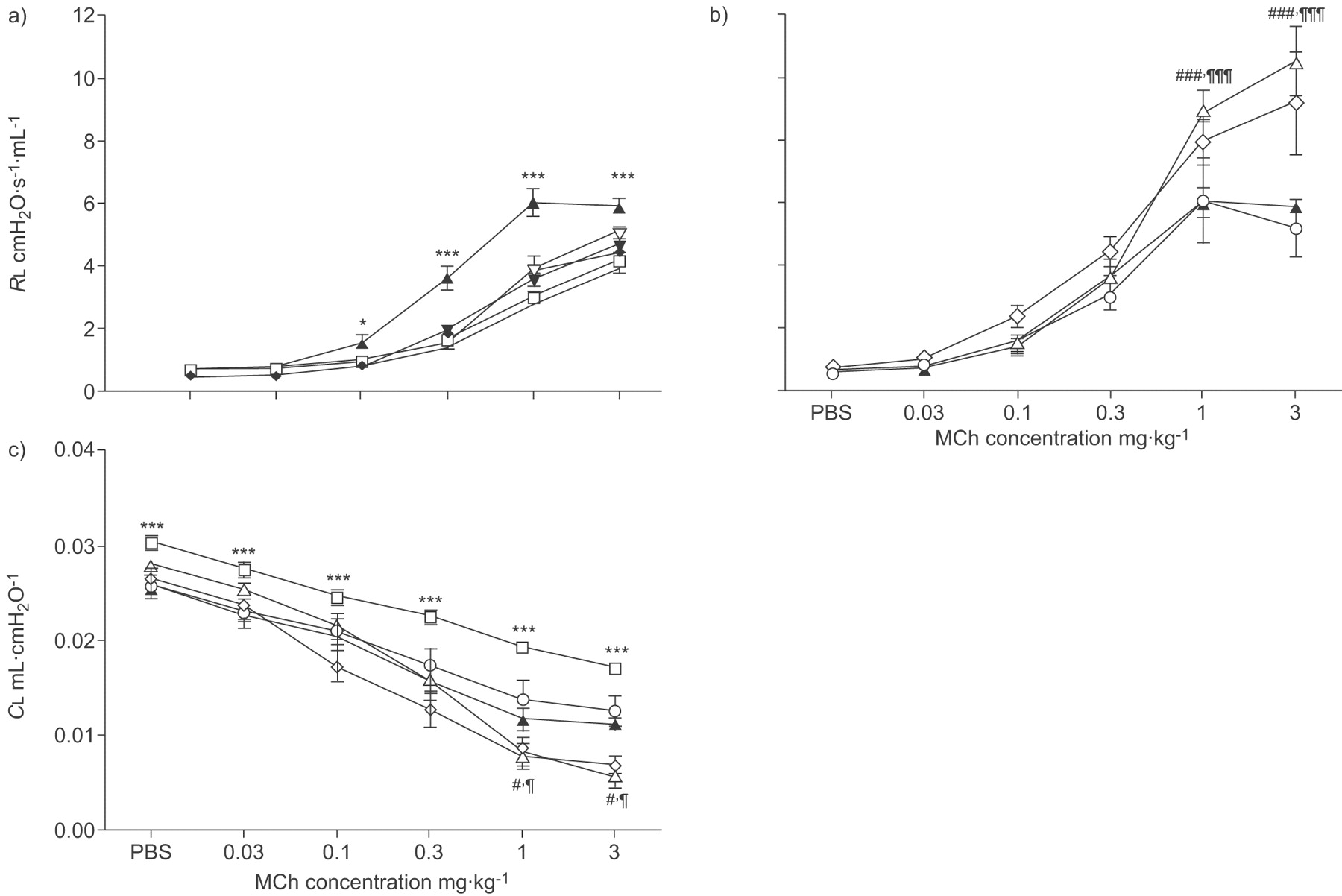
There were generally no differences in baseline RL and CL between the different treatments. In OVA, sensitised and challenged mice both the airway resistance (RL,max 5.9±0.2 cmH2O·s−1·mL−1; p<0.001) and the sensitivity (calculated as ED50 0.37±0.06 mg·kg−1 body weight; p<0.001) to tail-vein injection of MCh was substantially increased compared with PBS-challenged controls (4.1±0.2 cmH2O·s−1·mL−1 and 0.75±0.09 mg·kg−1 body weight, respectively; fig. 2a⇓; table 1⇓).

a and b) Lung resistance (RL) to methacholine (MCh) after ovalbumin (OVA) sensitisation and OVA or PBS challenge (OVA is similar in a and b). c) Pulmonary compliance (CL) to MCh after OVA sensitisation and OVA or PBS challenge. Drug interventions were administrated both during the OVA challenge and prior to the MCh challenge. Data points are represented as mean±sem. a) PBS (□): n = 12; PBS-diclofenac (♦): n = 6; PBS-FR122047 (▿): n = 6; PBS-lumiracoxib (▾): n = 6; OVA (▴): n = 12. b) OVA: n = 12; OVA-diclofenac (▵): n = 7; OVA-FR122047 (⋄): n = 9; OVA-lumiracoxib (○): n = 8. c) PBS: n = 12; OVA: n = 12; OVA-diclofenac: n = 7; OVA-FR122047: n = 9; OVA-lumiracoxib: n = 8. ***: p<0.001 and *: p<0.05 OVA compared with PBS-challenged controls and ###: p<0.001 and #: p<0.05 diclofenac; ¶¶¶: p<0.001 and ¶: p<0.05 FR122047 compared with OVA controls.
Measurement of airway function in anaesthetised mice following ovalbumin(OVA) or PBS challenge: the maximal reactivity and sensitivity to methacholine challenge
There were no changes in lung resistance or sensitivity in sensitised and PBS-challenged mice treated with either of the COX inhibitors compared with PBS controls alone (p>0.05; fig. 2a⇑).
Administration of the nonselective COX inhibitor diclofenac and the selective COX-1 inhibitor FR122047 during OVA challenge (fig. 1⇑) produced a significantly enhanced airway resistance compared with OVA sensitisation and challenge controls (p<0.001; fig. 2b⇑). Despite the marked increase in amplitude after treatment with diclofenac and FR122047, there was however no further change in sensitivity (p>0.05; table 1⇑). In contrast, the selective COX-2 inhibitor lumiracoxib did not change either the airway resistance or sensitivity compared with OVA-challenge controls (p>0.05; fig. 2b⇑; table 1⇑).
Lung compliance was measured to assess a second parameter of lung function. The sensitisation and challenge with OVA induced a decrease in CL (p<0.001; fig. 2c⇑) compared with the control mice. In mice treated with diclofenac and FR122047, but not lumiracoxib, the change in CL was further decreased (p<0.05; fig. 2c⇑), reflecting the altered RL with these treatments (fig. 2b⇑).
Cellular response in BAL
Compared with PBS controls, the total cell number and the number of eosinophils in BAL were markedly increased in OVA sensitised and challenged mice (p<0.001) as well as macrophages, neutrophils and lymphocytes (p<0.01, p<0.01 and p<0.001, respectively; fig. 3⇓). In mice treated with diclofenac or lumiracoxib, the total cell response was reduced (p<0.001) as well as the number of eosinophils (p<0.01 and p<0.001, respectively; fig. 3⇓). Treatment with FR122047 did not significantly alter the total cell response. In PBS-challenged control groups, diclofenac had no effect on total cell number or composition of the cells in the BAL fluid (fig. 3⇓).

a) Total cell count in bronchoalveolar lavage (BAL) fluid in ovalbumin (OVA) sensitised and OVA or PBS-challenged mice. b) Cellular composition in BAL. Drug interventions were administrated both during the OVA challenge and prior to methacholine challenge. Data are represented as mean±sem. a) PBS: n = 12; PBS-diclofenac: n = 6; OVA: n = 12; OVA-diclofenac: n = 8; OVA-FR122047: n = 12; OVA-lumiracoxib: n = 12. b) PBS: n = 12; PBS-diclofenac: n = 6; OVA: n = 11; OVA-diclofenac: n = 8; OVA-FR122047: n = 12; OVA-lumiracoxib: n = 10. □: PBS; ░: PBS diclofenac; ▪: OVA; ▓: OVA-diclofenac; ▒: OVA-FR122047; ┘: OVA-lumiracoxib. *: p<0.05; **: p<0.01; and ***: p<0.001 compared with PBS-challenged controls. ##: p<0.01 and ###: p<0.001 compared with OVA controls.
Levels of prostanoids and CysLTs in BAL
The levels of PGD2 (p<0.001; fig. 4a⇓), PGE2 (p<0.001; fig. 4b⇓), TXA2 (p<0.001; fig. 4c⇓) and CysLTs (p<0.01; fig. 4d⇓) increased significantly in BAL above basal levels in OVA-challenged mice. For the prostanoids, the ratio of release was PGE2 >>PGD2>TXA2 (21.8, 5.3 and 1, respectively).

Release of mediators in bronchoalveolar lavage (BAL) fluid in ovalbumin (OVA) sensitised and OVA or PBS-challenged mice (mean±sem). Concentration (pg·mL−1 BAL) of a) prostaglandin (PG) D2, b) PGE2, c) thromboxane (TXA) B2 and d) cysteinyl leukotrienes (CysLT). Drug interventions were administrated both during the OVA challenge and prior to methacholine challenge. *: p<0.05; **: p<0.01; and ***: p<0.001 compared with PBS-challenged controls. ##: p<0.01 and ###: p<0.001 compared with OVA controls.
Treatment with diclofenac or FR122047 significantly inhibited basal and OVA induced release of PGD2 (p<0.001; fig. 4a⇑), PGE2 (p<0.001; fig. 4b⇑) and TXA2 (p<0.001; fig. 4c⇑). Lumiracoxib attenuated the production of TXA2 (p<0.01; fig. 4c⇑) and there was a tendency for PGE2 inhibition, but it was not able to cause the same degree of inhibition of the release of PGD2 (p>0.05; fig. 4a⇑ and b). None of the three COX inhibitors had any effect on the CysLTs (p>0.05; fig. 4d⇑).
Levels of cytokines in BAL
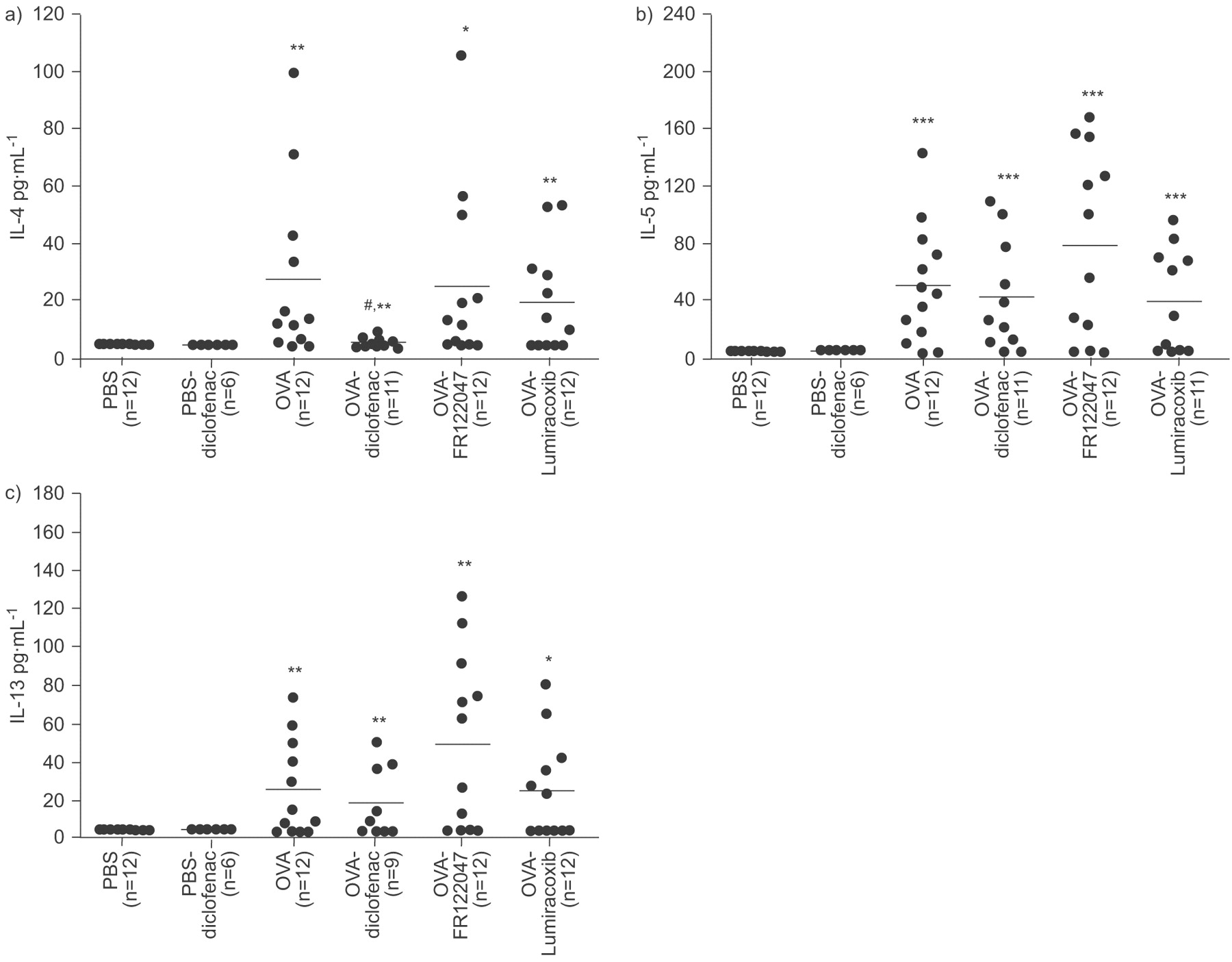
OVA challenge increased the BAL levels of IL-4 (5.5-fold; p<0.01; fig. 5a⇓), IL-5 (10.3-fold; p<0.001; fig. 5b⇓) and IL-13 (5.2-fold; p<0.01; fig. 5c⇓). Treatment with COX inhibitors generally induced no change in the level of cytokines in BAL fluid compared with OVA controls (fig. 5⇓), with the exception that mice treated with diclofenac had slightly reduced levels of IL-4 (four-fold; p<0.05) compared with OVA (fig. 5a⇓). In PBS-challenged control groups, diclofenac had no effect on the release of cytokines in the BAL fluid. There were no detectable levels of IL-10, TNF or IFN-γ in any of the groups.

Cytokine release in bronchoalveolar lavage (BAL) fluid in ovalbumin (OVA) sensitised and OVA or PBS-challenged mice. Levels of a) interleukin (IL)-4, b) IL-5 and c) IL-13. Drug interventions were administrated both during the OVA-challenge and prior to methacholine challenge. Results are expressed as points and mean. Each point represents results from one animal. *: p<0.05; **: p<0.01; and ***: p<0.001 compared with PBS-challenged controls and #: p<0.05 compared with OVA controls.
Intervention with COX inhibitors during either OVA or MCh challenge
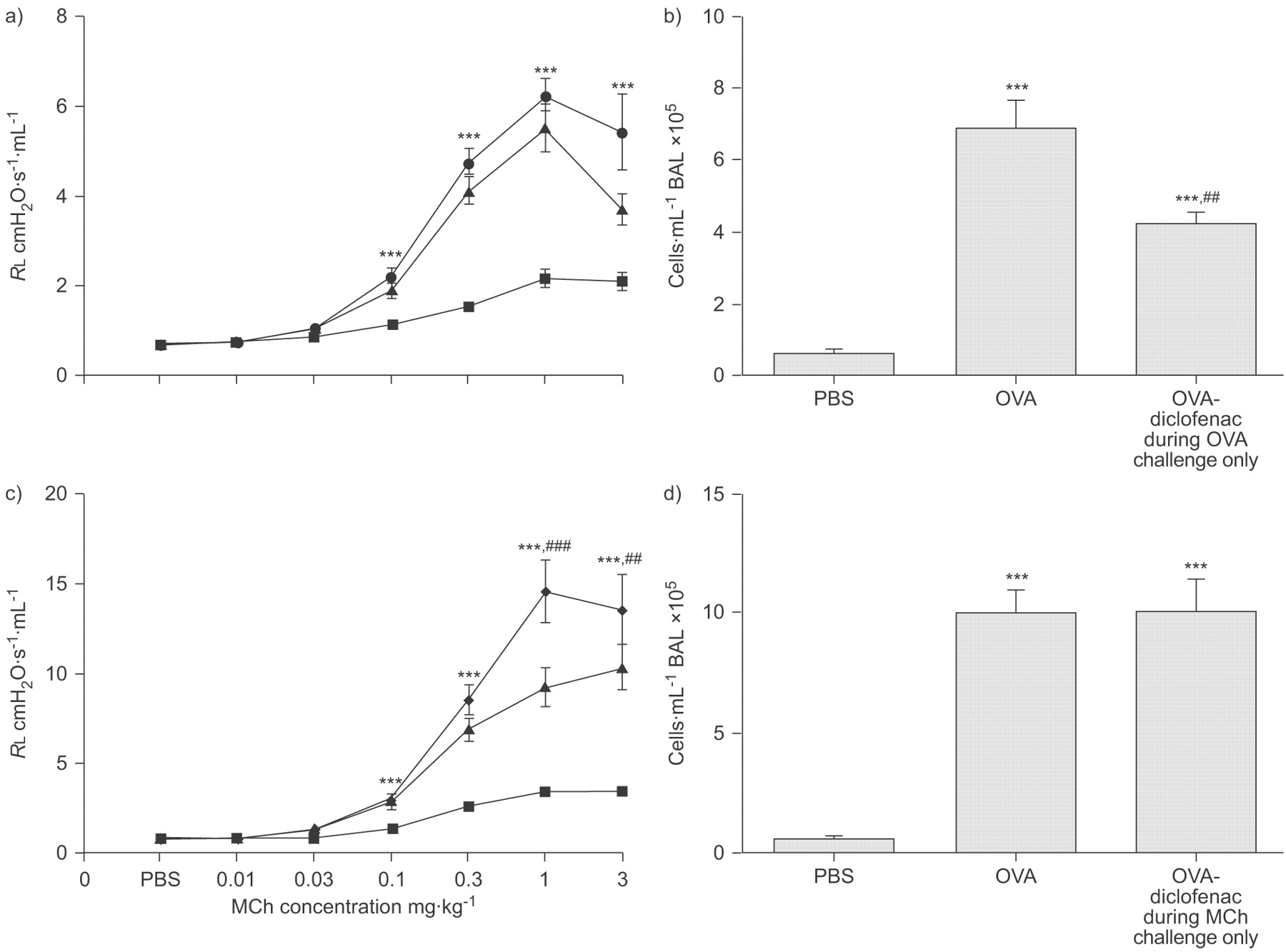
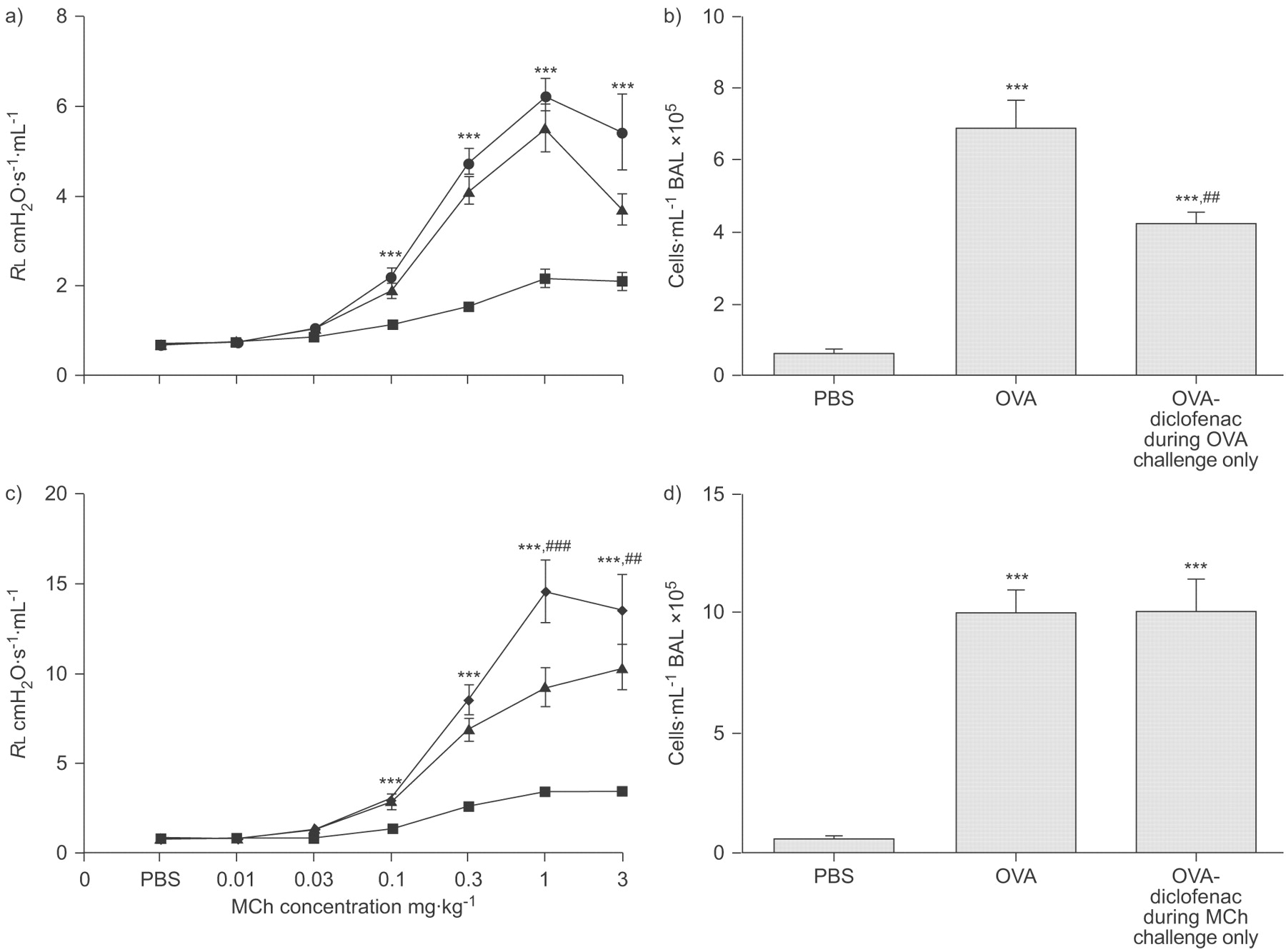
Administration of diclofenac only during OVA challenge did not further increase the resistance to MCh compared with OVA controls (fig. 6a⇓), but significantly reduced the cell response in BAL (p<0.01; fig. 6b⇓).
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Ovalbumin (OVA) sensitisation and challenge in mice treated with diclofenac a and b) only during OVA challenge or c and d) only during methacholine (MCh) challenge. a and c) Lung resistance (RL) to MCh and b and d) total cell count in bronchoalveolar lavage (BAL) fluid. Data points are represented as mean±sem. a) PBS (▪): n = 9; OVA (▴): n = 7; OVA-diclofenac during OVA-challenge only (•): n = 8. b) PBS: n = 10; OVA: n = 10; OVA-diclofenac: n = 9. c) PBS: n = 8; OVA: n = 9; OVA-diclofenac during MCh-challenge only (⧫): n = 9. d) PBS: n = 10; OVA: n = 10; OVA-diclofenac during MCh-challenge only: n = 10. ***: p<0.001 compared with PBS-challenged controls. ##: p<0.05; and ###: p<0.001 diclofenac compared with OVA controls.
When diclofenac was administered only prior to MCh challenge, the resistance to MCh was enhanced (p<0.01; fig. 6c⇑), compared with OVA controls. In contrast to animals that received diclofenac during OVA challenge (figs 3a⇑ and 6b⇑) there was no reduction in cell response in BAL (fig. 6d⇑).
DISCUSSION
The aim of the current study was to gain a more detailed understanding of the role of COX products in the eosinophilic airway reaction induced by OVA challenge in sensitised animals. It was discovered that there was a dissociation of the effects of COX inhibition during the OVA challenge on airway inflammation and AHR, suggesting that inflammatory cells in BAL do not change in parallel with AHR. Intervention with COX-inhibitors during the OVA challenge indicated that COX-1 activity predominantly generated prostanoids that are bronchoprotective and thus, serve to protect against further increases in AHR. In contrast, COX-2 activity was associated with infiltration of inflammatory cells in the lung, supporting a pro-inflammatory function of that pathway. Therefore, the findings also indicated distinct roles of prostanoids generated along the COX-1 and COX-2 pathways.
Further investigation of the time-point of action of COX inhibition, showed that the administration of diclofenac only during the OVA challenge reduced the cell response in BAL without any effect on the increased AHR to MCh induced by the allergen challenge (fig. 6a⇑ and b). In contrast, administration of diclofenac only prior to the MCh challenge enhanced allergen-induced AHR to MCh but did not affect cell response in BAL (fig. 6c⇑ and d). In mice sensitised but not challenged with OVA, COX inhibition did not affect RL or ED50 to MCh (fig. 2a⇑). This finding supports that the effect of COX inhibition was not a consequence of changes in baseline airway physiology but due to interference with a functional change that was caused by the OVA challenge.
Furthermore, OVA challenge increased AHR to MCh both with increased reactivity and sensitivity. Inhibition with diclofenac and FR122047, caused a further increase of reactivity without a change in sensitivity to MCh, whereas the selective COX-2 inhibition by lumiracoxib did not change the AHR induced by OVA alone. These findings (table 1⇑) suggest that the enhanced AHR is caused by COX-1 inhibition that it is due to an effect on smooth muscle hyperreactivity, whereas the increased AHR after OVA challenge compared with PBS controls includes additional changes that also affect the sensitivity to MCh 14. There is indeed data from studies in asthmatics to support primary modulation of airway smooth muscle reactivity as a distinct effect in AHR, induced for example by drug treatments 15, 16.
Our findings, implicating that the COX-1 pathway is critical for determination of AHR in this model, are consistent with the decreased AHR to MCh in mice deficient of COX-2 and over-expressing human COX-1 17. Likewise, studies in mice deficient of either COX-1 or COX-2 genes showed that only the allergic COX-1−/− mice exhibited increased airway reactivity to MCh 3, 4. Thus, PGs generated by COX-1 activity have a bronchoprotective role.
Mice treated with diclofenac and lumiracoxib during OVA challenge had decreased eosinophilic inflammation in BAL, whereas FR122047 did not affect the cellular response. This indicates that COX-2 generates prostanoids that mediate the accumulation of cells in the airways. It has been reported that mice deficient in COX-2 and over-expressing human COX-1, or animals with COX-inhibition before sensitisation, showed a similar or increased inflammatory response similar to OVA controls 1, 2, 17. In contrast, mice deficient in either COX-1 or COX-2 demonstrated an attenuated inflammation 4, again indicating a role for COX products to recruit inflammatory cells. Moreover, although a decrease of inflammation was seen in our experiments after COX-2 inhibition, the allergen-induced AHR was obtained to the same degree as in animals not given the inhibitor. This lends support to the concept of separate effects of prostanoids on airway inflammation and AHR.
Whereas the study established the different effects of iso-enzyme selective COX inhibitors on AHR and inflammation, it does not define the particular PG that mediated the effects of the inhibition. However, both PGD2 and PGE2 induce strong and potent relaxations of mice airway smooth muscle through activation of DP1 and EP2 receptors, respectively, 18–20, and COX-1 inhibition caused an attenuation of PGD2 and PGE2 in BAL. Therefore, it is likely that the enhancing effects of COX-1 inhibition on the increased AHR after OVA challenge may be explained in terms of removal of PGE2 and PGD2. Consistent with our study, Peebles and colleagues 1, 2 also found that COX inhibition caused a reduction of PGE2 levels together with an enhanced AHR. However, in their study, not only COX-1 but also COX-2 inhibition decreased PGE2, which may seem to be at variance with our results 1, 2. As they used a different COX-2 inhibitor 21 and also administered COX-inhibitors prior to sensitisation and continuously during the whole study 1, 2, the data are not directly comparable and further studies are required to resolve this issue.
In our study, the selective COX-2 inhibition by lumiracoxib was not able to cause the same degree of inhibition of PGE2 and PGD2 as COX-1 inhibition in BAL and it did not affect the OVA-induced AHR, further supporting the interpretation that these two PGs are likely to serve a bronchoprotective function. In line with our findings in this murine model, protective activity of PGE2 is also observed in asthmatics 22. Inhalation of PGE2 prevents allergen-induced early and late airway responses after allergen challenge 23–25. The effect of PGE2 on the airway responses to allergens may be explained both in terms of relaxation of the smooth muscles and by inhibition of the release of mast-cell mediators 26. However, as COX inhibition did not affect the allergen-evoked release of CysLT or cytokines in our study, it is concluded that smooth muscle relaxation was the predominant mechanism in this particular model.
It is acknowledged that our data do not define which PG mediated the COX-2 dependent effects on cellular inflammation. Local synergy in the tissue between vascular effects of PGE2 and chemotactic mediators 27, 28, or chemoattractant receptor-homologous molecule expressed on T-helper type (Th) 2 cell-mediated effects of PGD2 22, 29 are two possible explanations but further studies are required to resolve the mechanism.
Confirming previous reports 30, 31, the major Th2 cytokines, IL-4, IL-5 and IL-13, were increased in BAL after OVA sensitisation. Previous studies have linked the development of OVA-induced AHR in particular to increased IL-13 levels observed after inhibition of either COX-1 or COX-2 1, 2. This particular increase of IL-13 was documented 4 days before assessment of AHR. However, consistent with our findings, the level of IL-13 was no different from animals challenged with OVA in the absence of COX inhibition at the time of AHR measurements 1, 2. It is possible that changes in cytokine levels during OVA challenge in the presence of COX inhibition may have contributed to our results but we did not measure the cytokines during the OVA challenge period. Conversely, the observed changes in PGE2 and PGD2 levels in BAL, together with the finding that AHR also increased when the COX inhibitor was given only during the day when AHR was assessed, suggests that modulation of OVA-induced increase of PGE2 and PGD2 in airway smooth muscle is sufficient to explain the amplificating effect of COX inhibition on AHR.
In conclusion, mice treated with a selective COX-1 inhibitor had enhanced AHR but no change in the accumulation of inflammatory cells in the lung, whereas mice treated with a selective COX-2 inhibitor displayed a decrease of cells in the BAL but no change of AHR. The implications of separate functions for COX-1 and COX-2 products gained further support as treatment with the nonselective COX inhibitor diclofenac combined the effects seen with the two selective COX inhibitors. The combined effects of the unselective COX inhibitor diclofenac on AHR and BAL cells also supports the fact that doses of FR122047 and lumiracoxib used in this study were effective and selective. Taken together, the present study indicates that AHR and the airway inflammatory response are distinct, and at least in part, uncoupled events. Furthermore, there is also a difference in time for the development of the two separate reactions during allergen challenge. This is, in fact, in line with recent observations in subjects with asthma where BAL inflammation is not a predictive surrogate marker of AHR 32.
Support statement
The current study was supported by the Swedish Heart Lung Foundation, the Swedish Research Council, the Stockholm Country Council Research Funds (ALF) and Karolinska Institutet (all Stockholm, Sweden).
Statement of interest
A statement of interest for this study can be found at www.erj.ersjournals.com/misc/statements.dtl
Acknowledgments
The authors would like to thank A. Hesselgren (Dept of Medical Biochemistry and Biophysics) for animal care and her assistance, I. Delin (The National Institute of Environmental Medicine, Division of Physiology; both Karolinska Institutet, Stockholm, Sweden) for EIA consulting and L. Gold (Scireq, Montreal, QC, Canada) for technical FlexiVent® support.
- Received February 28, 2008.
- Accepted February 9, 2009.
- © ERS Journals Ltd
References










