





Abstract
Under steady state conditions the intracellular pathway is the major route of collagen catabolism in tissues characterised by rapid collagen turnover. In the lung, the collagen is subject to continuous remodelling and turnover however, the intracellular pathway of collagen degradation is unusual under physiological conditions.
The current authors previously described crystalloid inclusions in alveolar macrophages of mice with genetic emphysema at the time of septal disruption. Using an immunogold technique these inclusions were identified as collagen-derived products and related to intracytoplasmic collagen degradation. To examine whether a different degree of protease burden in lung interstitium may influence the route of intracellular collagen degradation, collagen phagocytosis by alveolar macrophages was studied in various mouse models of emphysema at the time when emphysema develops.
Evident collagen by-products in alveolar macrophages were observed in destructive processes characterising spontaneous models of emphysema either with negligible (blotchy mouse) or moderate (pallid mouse) elastase burden. On the other hand, intracellular collagen by-products were appreciated only in a few macrophages from tight-skin mice with high elastolytic burden and could not be observed in mice with a very severe burden after elastase instillation. In conclusion, the interstitial level of proteases burden can affect the way by which the collagen is cleared (intracellularly versus extracellularly).
- alveolar macrophages
- animal models of emphysema
- crystalloid inclusions
- fibrillar collagens
- intracellular collagen by-products
- protease burden
This work was supported by a grant from Ministero dell'Istruzione, Università e Ricerca Scientifica (MIUR; Rome, Italy) and by a grant from University of Siena (PAR, Progetti).
Collagenous proteins are major constituents of all extracellular matrices. Several types of collagen are recognised on the basis of variations in the structure of their polypeptide chains. Types I, II, III, V and XI collagens form fibrils and are hence known as fibrillar collagens 1.
A collagen network composed largely of type I and III fibrillar collagens is found in the extracellular space of the lung alveolar walls, skin, myocardium, uterus and several other tissues. It plays a dominant role in maintaining the tissue architecture and chamber geometry. Given its tensile strength, fibrillar collagen is a major determinant of tissue stiffness 1.
Under physiological conditions, synthesis and degradation of collagen is balanced to maintain tissue homeostasis 2, 3. In lung tissue, disrupted homeostasis leads to an imbalance between synthesis and degradation, resulting in either collagen destruction (i.e. emphysema) or excessive collagen deposition (i.e. fibrosis) 4, 5.
There are two important pathways for collagen degradation 6, 7. The extracellular pathway involves matrix metalloproteinases that are capable of degrading ≥1 types of collagen, while the intracellular route involves lysosomal degradation of fibrils internalised by cells such as macrophages, fibroblasts and osteoclasts. The mechanisms that regulate collagen phagocytosis in vivo are poorly defined although it is widely accepted that under steady state conditions the intracellular pathway is the major route of collagen catabolism in several tissues and organs characterised by rapid collagen turnover (periodontal ligament, gingiva, periosteum, uterus during postpartum involution, etc.) 8. In this context, and especially in soft connective tissues, fibroblasts appear to play a major role both in extracellular and in intracellular collagen degradation 8.
In the lung, the collagen is subject to continuous remodelling and turnover 3 however, the intracellular pathway of collagen degradation is not seen under physiological conditions.
The current authors recently reported a relevant collagen phagocytosis by lung macrophages during the ongoing destructive process that characterises the onset of spontaneous emphysema in pallid mice with α1‐proteinase inhibitor deficiency 9. In these animals, the present authors demonstrated the presence of intracytoplasmic crystalloid inclusions similar to those observed in macrophages from connective tissue undergoing rapid remodelling 10, 11, whose nature was for a long time an object of debate 12. Using electron microscopy and an immunogold-labelling technique, these inclusions were identified by the present authors as collagen-derived products and have been related to the intracellular route of collagen degradation that takes place during septal disruption.
Although knowledge of the role and mechanisms of the intracellular pathway of collagen digestion by macrophages has increased considerably during the past years 13, 14, many details of the processes involved still remain to be elucidated. Actually, in vivo studies are still lacking in this area. When investigating this field, questions such as which factors initiate or modulate collagen phagocytosis may be of importance in understanding the pathophysiology of interstitial lung diseases, such as emphysema and fibrosis.
In a recent study 15 it has been clearly demonstrated that macrophages can adhere to denatured but not to native type I and III collagens. This has been ascribed to specific surface receptors (macrophages scavenger receptors (MSRs)) that bind fibril-forming collagens which have been denatured. It is well known that during acute and chronic lung injury, extracellular matrix proteins are exposed to an environment laden with proteolytic enzymes and/or oxidants, which can degrade and/or alter their native conformation 16.
To examine whether the degree of protease burden in lung interstitium influences the route of intracellular collagen degradation, the current authors studied collagen phagocytosis by alveolar macrophages in various mouse models of emphysema at the time when emphysema develops. The mouse strains selected for this study develop spontaneous lung lesions in presence of negligible, moderate or high interstitial protease burden. The occurrence of intracytoplasmic crystalloid inclusions in alveolar macrophages was also studied in a mouse model of experimental emphysema in which a very severe interstitial protease burden is obtained via intratracheal instillation of human neutrophil elastase.
Materials and methods
Animals
Mice of the strain C57 Bl/6J (C57), and its mutants pallid C57 Bl/6J pa/pa and tight-skin C57 Bl/6J Tsk/pa mice were from the current authors' colony. Blotchy C3H Blo/+ mice were from the Department of Comparative Biology, Charing Cross and Westminster Medical School (London, UK). The mice were housed in groups of two to four in macrolon cages. Room temperature was kept at 22–24°C; relative humidity at 40–50%; food and water were supplied ad libitum.
The Local Ethical Committee of the University of Siena approved all animal experimentation.
Experimental protocol
Mice of various strains (tight-skin, blotchy, and pallid) known to develop spontaneous emphysema were used at the time of septal disruption. Thus, tight-skin mice were 1–2 months old 17, blotchy mice 3–6 months old 18 and pallid mice 8–12 months old 19. In an additional study, emphysema was induced in C57 mice known not to develop this lesion spontaneously. In particular, 2 month-old C57 mice received intratracheally, under light anaesthesia 25 µg of elastase from human sputum (Elastin Products, Co., Owensville, Missouri, USA) dissolved in 50 µL saline. These animals were used at 14–21 days after elastase instillation. The animal models that were used and the time of development of emphysematous lesions are entered in table 1⇓.
Schematic overview of the development of emphysema in different mouse models
At the selected time points, mice were anaesthetised with sodium pentobarbital and were killed by severing the abdominal aorta. After thoracotomy, the thoracic viscera were immediately removed. The lungs were washed in cold saline and then handled for morphological studies. Alternatively, they were used for bronchoalveolar lavage (BAL) determinations.
Morphological examination of pulmonary macrophages from lung sections
For morphological investigation, lung samples were fixed in 2% glutaraldehyde in 0.1 M sodium cacodylate buffer, pH 7.2 for 2 h at 4°C, postfixed in 1% buffered osmium tetraoxide (OsO4), dehydrated and embedded in epoxy resin (Araldite). Small cubes (1 mm3) of lung tissue taken at several levels from lungs of the various strains of mice were processed in a similar way. Lung semithin sections (0.5–1 µm thick) were cut on a LKB Ultratome V and stained with 1% toluidine blue. Ultrathin (∼600 Å) sections from lung specimens were mounted on copper grids, stained with uranyl acetate-lead citrate and examined in a Philips 300 electron microscope.
Determination of elastase burden by immuno-electron microscopy
The immunogold method (postembedding technique) was used to localise elastase in thin lung sections prepared for electron microscopy using antimouse leukocyte elastase (anti‐MLE) antibodies as previously described in detail 20. Briefly, lung tissue blocks (five per animal) taken from five tight-skin, five blotchy and five pallid mice were used. The density of gold particles per square micrometer of lung tissue was determined for each of the micrographs with a superimposed quadratic lattice grid. A total of 50 micrographs were thus analysed for each animal, and the average gold particle density of lung connective tissue for the two groups was calculated.
The elastolytic burden was scored as negligible (−: <2 gold particles per µm2), moderate (+: 2–8 gold particles per µm2) and marked (++: >8 gold particles per µm2). The elastolytic burden of the C57 Bl/6J mice could not be determined since these mice were injected with human neutrophil elastase that does not react with anti-MLE antibodies. However, the amount of human neutrophil elastase injected intratracheally allows the elastolytic burden in these mice to be considered “very severe”.
Immunolocalisation of collagen in alveolar macrophages
Some tissue samples, handled in a similar way as reported above to localise elastase, were used to investigate the nature of crystalloid inclusion observed in the different animal groups. Collagen by-products in pulmonary macrophages were localised by an immunogold method using rabbit antimouse collagen I immunoglobulin (Ig)G or rabbit antimouse collagen III IgG, as previously reported 9.
Bronchoalveolar lavage and biochemical analysis
After sacrificing animals, the trachea was isolated and then cannulated with a 20 gauge blunt needle. By the aid of a peristaltic pump (P‐1 Pharmacia, Uppsala, Sweden) the lungs were repeteadly lavaged three times with 0.6 mL normal saline. The average recovery was 95%. The lavage fluid was then centrifuged to remove cells and supernatants were assayed for hydroxyproline concentration 22 and lactic dehydrogenase (LDH) 23 activity. Enzyme activity was reported in international units (IU) which are equal to micromoles of substrate converted per minute.
Statistical analysis
For each parameter either measured or calculated, the values of the individual animals were averaged and the SD was calculated. The significance of the differences was calculated using one way analysis of variance (F‐test). A p‐value of <0.05 was considered significant.
Results
The animal models used in this study, and the different time of development of emphysematous lesions are entered in table 1⇑.
Intracytoplasmic crystalloid inclusions were found in alveolar macrophages of the three strains of mice which spontaneously develop emphysema: blotchy (fig. 1a⇓), tight-skin (fig. 1b⇓) and pallid (fig. 1c, 1d⇓). These structures were morphological similar in all strains. In particular, at high magnification (fig. 2a⇓) the inclusions had a “needle-like” appearance, were electron dense and were bounded by a single membrane. In general, macrophages containing inclusions were endowed with an active membrane, as evinced by the presence of fine pseudopodia and an irregular outline. In some alveoli, the crystalloid “needles” appeared as free structures associated with cellular debris.

a) Electron micrograph of an alveolar septum of a 5 month-old blotchy mouse showing an alveolar macrophage containing crystalloid inclusions. Details of these inclusions are shown in fig. 2a⇓. b) Electron micrograph of an alveolar macrophage from a 1 month-old tight-skin mouse. Some crystalloid inclusions are present in the cytoplasm. c) Alveolar macrophage from an 8 month-old pallid mouse containing some crystalloid inclusions bound by a single membrane. d) Alveolar macrophage from a 12 month-old pallid mouse. Several crystalloid inclusions are evident. e) Alveolar macrophage from a 12 month-old C57 Bl/6J mouse. No crystalloid inclusions can be seen. f) Alveolar macrophage from an elastase-treated C57 Bl/6J mouse. The cytoplasm is free from crystalloid inclusions. Sections were stained with uranyl acetate and lead citrate. Scale bar=1 µm.

a) Detail of fig. 1a⇑ showing crystalloid inclusions bound by a single membrane. b) Alveolar macrophage from a 5 month-old blotchy mouse showing a positive reaction for collagen type I antibody on the crystalloid inclusions. Inset shows a lower magnification of these structures. c) Alveolar macrophage from a 5 month-old blotchy mouse showing a positive reaction for collagen type III antibody on the crystalloid inclusions. Inset shows a lower magnification of these structures. Sections were stained with uranyl acetate and lead citrate. Scale bar=2 µm.
Additionally, as previously reported for the pallid mouse, the inclusions observed in the blotchy and tight-skin mice showed a positive reaction for collagen type I (fig. 2b⇑) as well as for collagen type III (fig. 2c⇑), by using an immunogold-labelling technique.
Contrary to these findings observed in strains that spontaneously develop emphysema, the crystalloid inclusions were never seen in alveolar macrophages from C57 Bl/6J (C57) mice from 2–16 months of age (fig. 1e⇑) 9. Similarly, no inclusions were found in macrophages from 2 month-old C57 in which emphysema was induced by intratracheal instillation of elastase (fig. 1f⇑).
The crystalloid inclusions could also be seen at light microscopy, in plastic sections stained with toluidine blue as dark thin bodies engulfing the cytoplasm of pulmonary macrophages from blotchy, pallid and tight-skin mice (fig. 3a–c⇓). These features were never observed in the cytoplasm of macrophages from C57 mice treated with elastase (fig. 3d⇓).

Semithin sections stained with toluidine blue from lungs of a) blotchy, b) pallid, c) tight-skin and d) elastase-treated C57 mice. Arrowheads indicate alveolar macrophages containing dark thin bodies. Insets show a higher magnification of these macrophages. Scale bar=7.8 µm.
The percentage of macrophages exhibiting “needle-like” inclusions is presented in table 2⇓. The pallid mice had the highest percentages of macrophages with inclusions (35–85%). At 12 months of age the vast majority of pallid macrophages (up to 85% of the total cell population) were completely filled with crystalloids. The blotchy mouse showed 10–20% of macrophages with inclusions, while the tight-skin mouse showed such changes only in very few macrophages (1–3%).
Elastolytic burden and distribution of macrophages with inclusions in different animal models of emphysema
The values of the elastolytic burden (reported as gold particles density) detected in animals developing spontaneous emphysema are also entered in the table 2⇑. The mean values of particles was 4.9±1.2 per µm2 in pallid mice and 9.6±1.6 per µm2 in tight-skin mice (p<0.05). These values are related to pallid mice of 8 months of age and tight-skin mice of 1 month of age, i.e. at the time when these mice developed emphysema and crystalloid inclusions first appeared. The level of gold particle density found within the alveolar wall of congenic C57 BL76J at the various ages (1–12 months) was not greater than the background levels detected on control lung sections incubated without primary antibody (0.7±0.8 per µm2).
Of interest, after elastase instillation no macrophages with inclusions could be detected in C57 mice in which the elastolytic burden could be assumed as “very severe”. In pallid mice with a lower elastolytic burden than tight-skin mice, a greater percentage of macrophages with crystalloid inclusions were detected than in the tight-skin mice. In blotchy mice in which the cause of emphysema cannot be the result of an elastolytic process, the origin of the intracytoplasmic crystalloid inclusions is probably related to the destruction of alveolar septa due to the connective tissue abnormalities of this mutant.
As mentioned above, the crystalloid inclusions begin to appear in macrophages of animals with spontaneous emphysema at the time in which the septal destruction takes place. In particular, they appeared in 8 month-old pallid and 4 month-old blotchy mice when a significant increase in LDH activities (+36 and 41%, respectively) in respect to average control values (94.11 and 86.43 IU·L−1) was observed in BAL fluids. At these times, detectable amounts of hydroxyproline were found in (BAL) fluids from pallid (0.170±0.015 µg·mL−1) and from blotchy (0.190±0.026 µg·mL−1) mice.
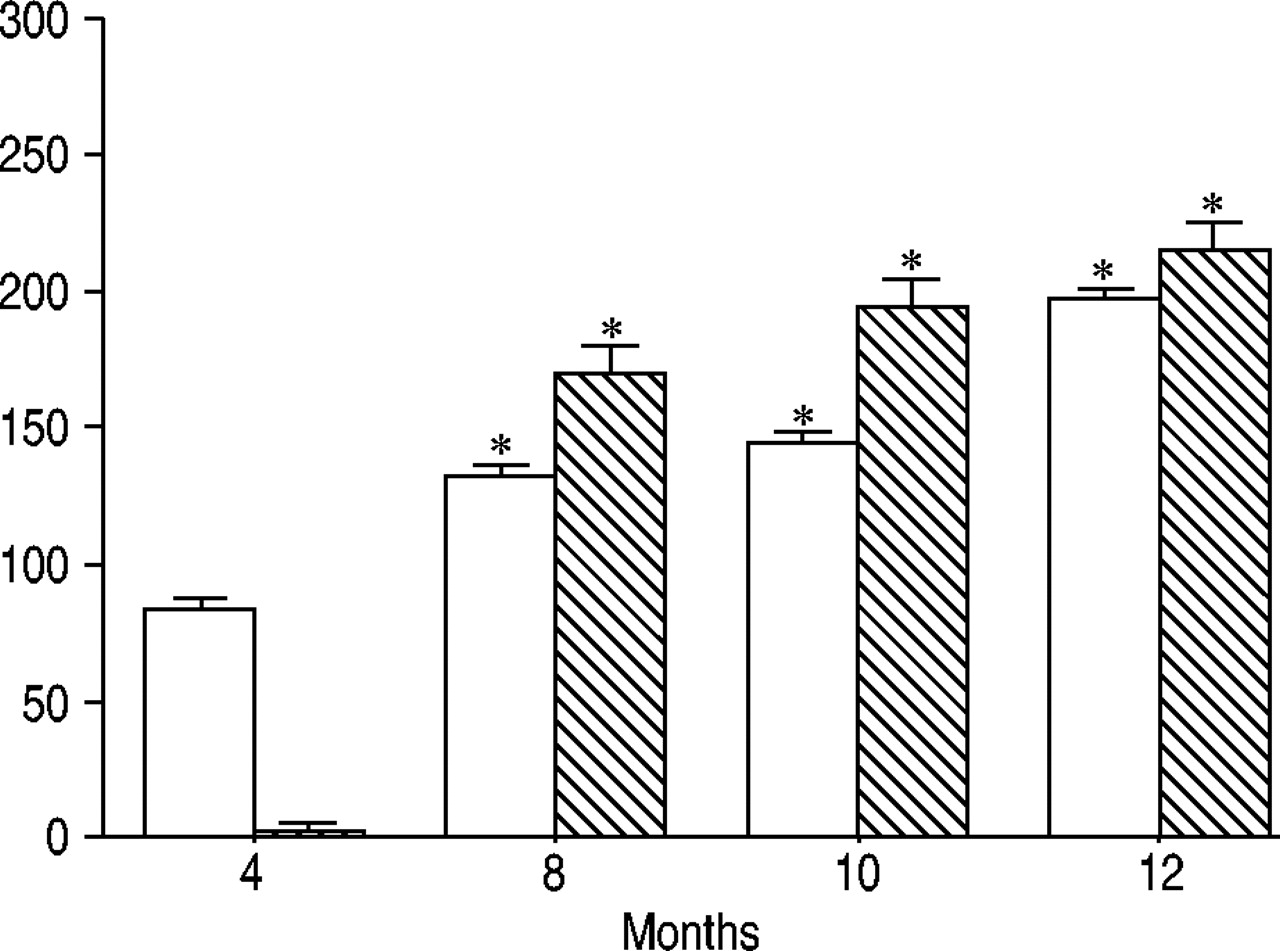
Of interest, the increase of the percentage of macrophages with the “needle-like” inclusions observed in pallid mice between 8 and 12 months of age, was accompanied by a progressive increase in the number of crystalloid inclusions within the cells (fig. 4⇓). The latter figure paralleled the increase of LDH activity and hydroxyproline content in BAL fluids (fig. 5⇓).

Percentage distribution of pallid macrophages containing different amounts of crystalloids at various time points. Macrophages have been scored according to the number of inclusions present in the cytoplasm (□: 0; ┘: <10;  : 10–40; ▪: >40).
: 10–40; ▪: >40).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Lactic dehydrogenase activity (□; IU·L−1) and hydroxyproline (┘; ng·mL−1) in bronchoalveolar lavage fluids from pallid mice at various ages. Values are the mean±sd of eight animals in each group. *: p<0.05 versus 4 month-old mice.
Discussion
The current paper reports an evident collagen phagocytosis by alveolar macrophages in the ongoing destructive processes that characterise two spontaneous models of emphysema withnegligible (blotchy) or moderate (pallid) elastase burden. On the other hand, intracellular collagen degradation was appreciated only in very few macrophages from tight-skin mice with a marked elastolytic burden and could not be observed in C57 mice in which an intratracheal elastase instillation results in a very severe protease burden.
The data presented suggest that the interstitial levels of protease burden can influence the way in which collagen is cleared. It is widely accepted that extracellular matrix (ECM) proteins, including type I and III collagens, are attacked by a variety of proteases, which acting alone or together, either completely digest the proteins or alter their native conformation 24–26. Under the different conditions of septal injury reported by the current authors, the local ECM proteins are exposed to various amounts of proteolytic enzymes that can degrade their molecules differently.
Elastolytic enzymes, and in particular elastases, are able to cleave interstitial collagens 24, 27. They can also promote collagen catabolism by modifying the protease-antiprotease balance in the interstitial environment either activating several metalloproteases with collagenolytic activity 28 or binding and/or digesting common inhibitors (i.e. α2‐macroglobulin, tissue inhibitors of metalloproteases) 29.
The ability of macrophages to differentially adhere via MSRs to proteolytically modified collagen type I and III has been recently demonstrated 15. This may represent an important factor in initiating collagen phagocytosis by macrophages, and may offer an explanation why the intracellular route ofcollagen degradation is not usual in the lung under physiological conditions.
The data obtained also support the idea that collagen phagocytosis by macrophages participates in the removal of injured extracellular matrix during the development of a slow progressive emphysema such as that occurring in pallid and blotchy mice. In fact, the crystalloid inclusions appeared only when a significant increase in LDH activity and detectable amounts of hydroxyproline could be demonstrated in BAL fluids. The progressive increase of the percentage of macrophages with crystalloid inclusions and of the number of these inclusions within the cells that were observed in pallid mice during the exacerbation of the ongoing destructive process (from 8 months of age onward) represent an additional support for such a view.
Of interest, in the aforementioned models of emphysema characterised by high protease burden (i.e. tight-skin and C57 mice after elastase treatment) the alveolar destruction is followed by an enhanced collagen synthesis. This results in marked deposition of collagen fibrils in the residual septa 30–33.
It has been proposed that lung synthesis of collagen and elastin can be controlled by the state of the extracellular matrix and peptides made soluble by elastase. Actually, there is evidence in vitro 34, 35 and in vivo 36 that products of collagen and elastin degradation by elastase may have a role in the deposition of these interstitial proteins. In the present authors' opinion the different routes of collagen degradation, the levels of protease burden and thereby the differences in cleavage and removal of collagen molecules may affect the remodelling of the interstitial matrix after an alveolar injury. The current authors are actively pursuing this hypothesis.
In summary, the present study provides evidence that different interstitial levels of protease burden in emphysema may be associated with different routes of collagen clearance (intracellular versus extracellular). Additionally, it may form the basis for future investigations regarding the remodelling of the interstitial matrix in emphysema.
- Received April 30, 2003.
- Accepted June 28, 2003.
- © ERS Journals Ltd
References









