





Abstract
Cellular senescence is a state of irreversible growth arrest. In this paper the authors examined whether bleomycin, an agent that causes pulmonary fibrosis, induces the senescence of alveolar epithelial cells.
Type II-like alveolar epithelial (A549) cells or rat primary type II cells were exposed to bleomycin and then evaluated for markers of cellular senescence. Bleomycin was also administered intratracheally in C57BL/6 mice.
The authors found that exposure to bleomycin induced cellular senescence in A549 cells and rat primary type II cells. The senescence was characterised by a dose- and time-dependent increase in senescence-associated β‐galactosidase activity, senescence-associated changes in cell morphology, an increase in cell size and lysosomal mass, the overexpression of p21 protein, and irreversible growth arrest. The intratracheal injection of bleomycin in mice induced an increase in senescence-associated β‐galactosidase activity in type II epithelial cells, reaching a maximum at day 7.
These results suggest that bleomycin induces a phenotype that is indistinguishable from that of senescence in alveolar epithelial cells. The induction of epithelial senescence by bleomycin may contribute to the pathway of impaired re-epithelialisation leading to pulmonary fibrosis.
Pulmonary fibrosis is characterised by sequential acute lung injury with progressive fibrosis and architectural distortion 1. The original hypothesis for the pathogenesis of pulmonary fibrosis was that chronic inflammation stimulates fibroblasts 2, 3. However, a newer hypothesis proposes that epithelial injury and impaired regeneration are sufficient to stimulate fibroblasts, without the need for ongoing inflammation 4. This hypothesis suggests that impaired re-epithelialisation, following lung injury, triggers the process of pulmonary fibrosis. An important pathological feature in patients with pulmonary fibrosis is a marked loss of type I cells without appropriate re-epithelialisation, while hyperplasia of abnormal type II cells occurs in other areas 4–7. The absence of re-epithelialisation is thought to induce the migration of activated fibroblasts, through defects in the epithelium from the interstitial to the intraluminal compartments, leading to pulmonary fibrosis 7. However, the mechanism of the impaired re-epithelialisation in pulmonary fibrosis is not well understood.
Cellular senescence is a state where cells can no longer divide, despite the presence of appropriate growth factors 8. This state can be induced either by telomere shortening (replicative senescence) or by telomere-independent signals, such as deoxyribonucleic acid (DNA) damage and oxidative stress (stress-induced senescence). The state of cellular senescence is associated with the following: irreversible growth arrest; a distinct flat, and enlarged cell morphology; an increase in senescence-associated β‐galactosidase (SA β‐gal) activity; andthe expression of cell cycle inhibitors, such as p16INK4a and p21CIP1/WAF1/Sdi1 8–10. Ample evidence indicates cellular senescence is important for phenotypic aspects of aging, such as delayed wound healing, a reduced immune response, and thinning of the skin 11. Recent studies have also indicated that cellular senescence plays an important role in the pathogenesis of chronic diseases, such as liver cirrhosis and atherosclerosis 12, 13. However, the role of cellular senescence in pulmonary fibrosis has not been examined. The authors hypothesised that when epithelial cells reach the senescence stage, epithelial regeneration ceases and fibrosis is promoted. In the present study, the authors examined whether bleomycin, an agent that causes pulmonary fibrosis, induces the senescence of alveolar epithelial cells.
Materials and methods
Animal treatment
The animal protocol was approved by the Animal Care and Use Committee of Tokyo Women's Medical University. An animal model for bleomycin-induced pulmonary fibrosis was generated, as described previously 14. The mice were anaesthetised with an intraperitoneal injection of sodium pentobarbital (Abbott Lab., North Chicago, IL, USA) followed by the intratracheal administration of 50 µL of a phosphate-buffered saline (PBS) solution containing bleomycin hydrochloride (5 mg·Kg−1 body weight; Nippon Kayaku, Co., Tokyo, Japan). At days 1, 4, 7, and 21 after the bleomycin injection, the mice were intraperitoneally treated with 30 µg of bromodeoxyuridine (BrdU); 4 h later, they were sacrificed using an intraperitoneal injection of a pentobarbital overdose. The lungs were removed, inflated by manual instillation with 50% OCT compound (Tissue-Tek®; Sakura Finetechnical Co. Ltd, Tokyo, Japan) quickly frozen, and sectioned (3 µm).
Cell cultures and bleomycin treatment
The alveolar type II-like epithelial cell line, A549 (ATCC #CCL‐185), was maintained in Dulbecco's modified Eagle's medium (DMEM) containing 10% foetal calf serum (FCS). The cells (104 cells·cm−2) between passages 27 and 35 were plated onto 8‐well-chamber slides or 35 or 60 mm tissue culture plates precoated with type I collagen (VitrogenTM, Cohesion, Palo Alto, CA, USA) and grown in the above medium. Once they reached 50% confluence, the cells were rinsed three times with PBS and incubated with DMEM containing 0.1% FCS with or without bleomycin (0.1–50 µg·mL−1) for the indicated periods.
Primary rat type II alveolar epithelial cells were isolated from adult male Sprague-Dawley rats, weighing 250 g, and maintained in DMEM containing 20% FCS, as previously described 15. The primary cells were studied at day 2 of the culture, a time at which they are type II cell-like as judged by accepted morphological and biochemical criteria 15. The primary cell preparations were of >90% purity as assessed by modified papanicolaou staining and >90% viability as assessed by trypan blue staining. The type II cells were exposed to bleomycin in DMEM containing 20% FCS because serum depletion killed the cells.
Cell proliferation analysis
After 120 h of bleomycin treatment, the A549 cells in the 8‐well-chamber slides were rinsed three times with PBS andincubated with DMEM containing either 0.1% FCS or10% FCS. After 48 h, the cells were pulse-labelled with 10 µM of BrdU for 60 min, the cells that had incorporated BrdU were detected using immunocytochemistry, as described below.
Cell size analysis
The cells in the 8‐well-chamber slides were fixed with 3% paraformaldehyde and stained with an eosin solution. Digitised video images of the cells were captured using an Olympus BX60 microscope (Olympus Optical Co., Ltd, Tokyo, Japan) with an Olympus DP50 CCD camera, and sentto an Olympus imaging microscopic workstation (CUSL2G40; Olympus Optical Co., Ltd) equipped with a computer running Microsoft Windows 98 and a computerised color image analysis software system (Win Roof Version 3.5, Mitani Corporation, Fukui, Japan). Each image was then subjected to operations that measured the cell size. The sizes of 300 cells from one well were averaged.
Lysosomal mass analysis
Morphological examination of the lysosomal mass was performed as previously described 16. Cells in the 8‐well-chamber slides were pretreated with 3×10−7 M of bafilomycin A1 (Sigma, St. Louis, MO, USA) or the vehicle dimethyl sulfoxide alone for 15 min at 37°C and then incubated with 5 µg·mL−1 of acridine orange (Sigma) for 10 min. After washing with PBS, the coverslips were mounted on slides and viewed immediately under an Olympus BX60 microscope using an Olympus longpass filter set comprised of a bandpass 450–490 nm exciter, a 510 nm dichroic mirror, and a long pass 520 nm emitter.
Senescence-associated β‐galactosidase
SA β‐gal staining was performed according to a previously described method 17, with slight modifications. Cell samples on 8‐well-chamber slides were fixed with 2% formaldehyde and 0.2% glutaraldehyde in PBS for 5 min at room temperature. Frozen lung tissue sections were fixed with 0.2% glutaraldehyde in PBS for 5 h at 4°C. The slides were rinsed with PBS and incubated with a SA β‐gal staining solution containing 40 mM sodium citrate (pH 6.0), 150 mM sodium chloride, 5 mM potassium ferricyanide, 5 mM of potassium ferrocyanide, 2 mM magnesium chloride, and 1 mg·mL−1 of (5‐bromo‐4‐chloro‐3‐indoyl β‐D galactoside (X-gal)). Some of the slides were then immunostained as described below.
An assessment of SA β‐gal intensity in relation to fibrosis was performed semi-quantitatively using lung tissue sections obtained from mice at 21 days after bleomycin treatment. For each slide, five fields of the fibrotic area and five fields of the nonfibrotic area were selected. For each field, the SA β‐gal intensity was graded: 0=no staining, 1=weak staining, 2=moderate staining, and 3=intense staining. The average grade of SA β‐gal intensity was obtained for both the fibrotic and nonfibrotic areas.
Immunohistochemistry and immunocytochemistry
After SA β‐gal staining, tissue sections were processed for routine immunohistochemistry. The primary antibodies were rabbit polyclonal antilaminin (1:50 dilution; Sigma), mouse monoclonal antipancytokeratin (1:100 dilution; Sigma), rabbit polyclonal antisurfactant protein A (SP‐A; 1:100, Santa Cruz Biotechnology Inc., Santa Cruz, CA, USA), rat monoclonal antimouse CD31 (1:25, Research Diagnostics Inc., Flanders, NJ, USA), rat monoclonal antimouse CD45 (1:100, Pharmingen, San Diego, CA), and mouse monoclonal anti-BrdU (1:100, Chemicon International, Temecula, CA, USA). For immunostaining with anti-BrdU, the slides were pretreated with 3 N HCl for 30 min and neutralised with 0.1 M borax buffer, pH 8.5, for 10 min. The primary antibody was reacted with biotinylated anti-IgG conjugated with horseradish peroxidase (HRP)‐labelled polymer (EnVison+ peroxidase; DAKO Japan, Kyoto, Japan). Immunoreactants were visualised by 3,3′‐diaminobenzidine and counterstained with a nuclear fast red solution. For the immunocytochemistry, cell samples on 8‐well-chamber slides were fixed in 70% ethanol at −20°C for 10 min and incubated with 0.3% H2O2 for 5 min, followed by incubation with 3% bovine serum albumin in PBS for 30 min. The slides were treated with 3 N HCl for 30 min, neutralised with 0.1 M borax buffer, pH 8.5, for 10 min, reacted with anti-BrdU antibody, and processed as above.
Tissue sections double-stained for SA β‐gal activity and either SP‐A, pancytokeratin, or CD31 antigen were assessed semiquantitatively. For each slide, 10 microscopic fields were randomly selected. For each field, the number of cells positive for each antigen and the number of cells positive for each antigen with SA β‐gal activity were counted. The average percentage of cells positive for each antigen and the SA β‐gal activity in the population of cells positive for each antigen was calculated.
Immunoblot analysis
Cell lysates were fractionated by sodium docecylsulfate-polyacrylamide gel electrophoresis (SDS-PAGE), transferred to polyvinylidene difluoride membrane, and probed using mouse monoclonal anti-p21 antibody (1:400; Santa Cruz) or rabbit polyclonal anti-actin (1:2,500; Sigma). The primary antibodies were detected using a horseradish peroxidase (HRP)-conjugated antibody, which in turn was visualised byenhanced chemiluminescence (SuperSignalTM, Pierce, Rockford, IL, USA).
Statistical analysis
All data are expressed as the mean±sem. Differences were examined for significance using an analysis of variance and the Scheffe test or with a two-tailed, unpaired t‐test, as appropriate.
Results
Treatment of A549 cells and primary rat type II cells with bleomycin induces cellular senescence
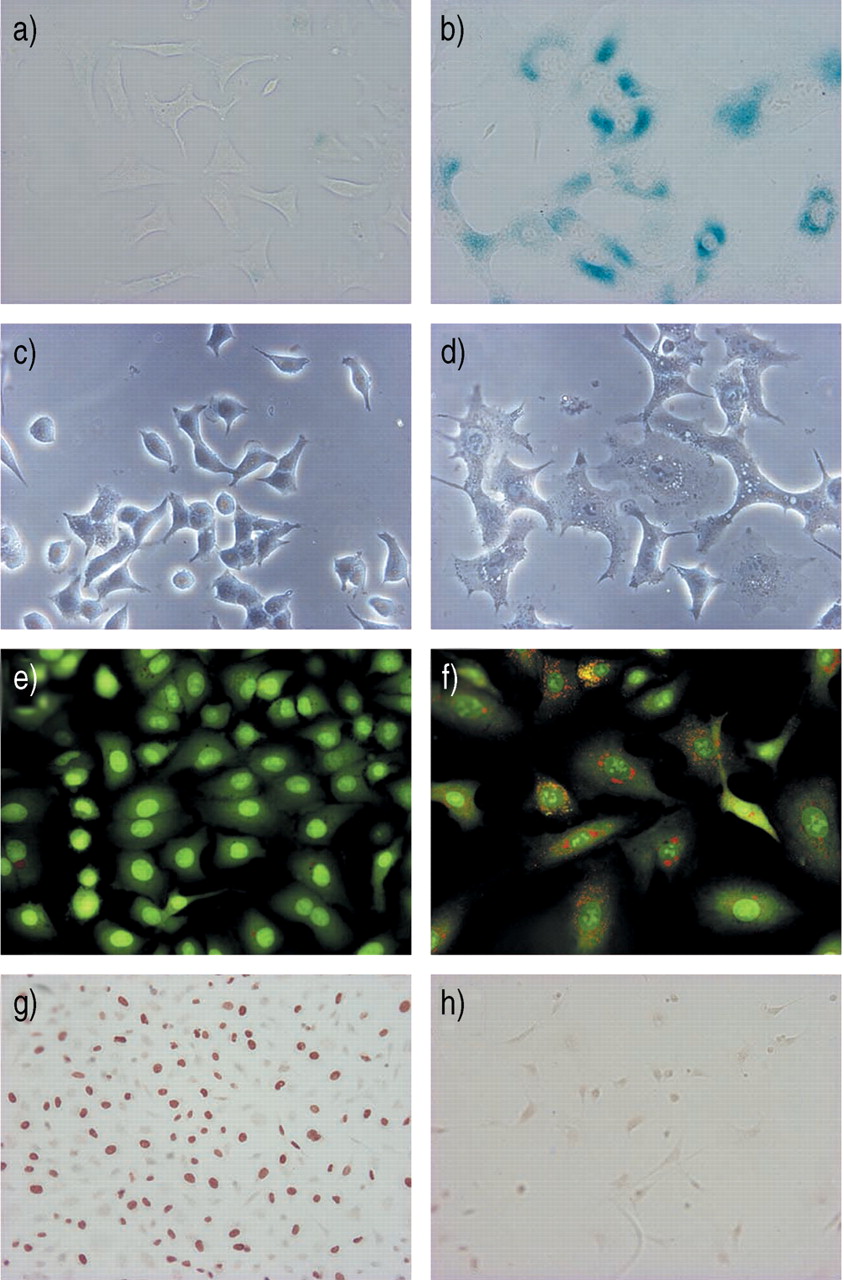
To determine whether bleomycin induces cellular senescence in lung epithelial cells, the authors treated alveolar type II-like epithelial (A549) cells with various concentrations ofbleomycin. Because bleomycin at concentrations of 100 µg·mL−1 or higher caused the death of >20% of the A549 cells, as determined by trypan blue staining, the authors treated the cells with bleomycin at sublethal concentrations of ≤50 µg·mL−1. When A549 cells were treated with 50 µg·mL−1 of bleomycin for 120 h they exhibited a phenotype that is typical of cellular senescence, as judged by an increase in SA β‐gal activity (fig. 1b⇓); a distinct, flat, and enlarged morphology (fig. 1d⇓); an increase in lysosomal mass (fig. 1f⇓); and irreversible growth inhibition (fig. 1h⇓) 8, 16–18. Quantitative analyses for cellular senescence showed that the treatment of A549 cells with bleomycin caused a dose-dependent and a time-dependent increase in SA β‐gal activity (fig. 2a and b⇓), a five-fold increase in cell size (fig. 2c⇓), and a dose-dependent inhibition of cell growth (fig. 2d⇓). As shown in figure 2b,⇓>95% of the A549 cells expressed SA β‐gal activity after treatment with 50 µg·mL−1 of bleomycin for 120 h. An immunoblot analysis also showed that the treatment of A549 cells with bleomycin caused a dose-dependent increase in the expression of p21CIP1/WAF1/Sdi1 protein, a cyclin-dependent kinase inhibitor that mediates cellular senescence in many types of cells (fig. 3⇓) 9, 19. The A549 cell is a lung cancer cell line which may not reflect the senescence property of normal alveolar epithelial cells. However, treatment of rat primary type II cells with bleomycin also caused an increase in SA β‐gal activity and a flat and enlarged morphology (fig. 4⇓), suggesting that bleomycin induces senescence of primary type II cells. However, the authors do not completely exclude the possibility that the senescent phenotype of alveolar epithelial cells after bleomycin treatment reflects a change from the type II to type I phenotype. Taken together, these results indicate that treatment with bleomycin induces a phenotype that is indistinguishable from that of cellular senescence in alveolar epithelial cells.

Senescent phenotype of A549 cells treated with bleomycin. A549 cells were treated without (a, c, e, and g) or with bleomycin (50 µg·mL−1) (b, d, f, and h) for 120 h and examined for markers of cellular senescence. Staining for senescence-associated β‐galactosidase activity (a, b). Cell morphology (c, d) characterised by flat, enlarged cells, after treatment with bleomycin (d). Lysosome staining with acridine orange (e, f) shows a marked increase in the number and size of lysosomes (orange fluorescence) in cells after treatment with bleomycin (f). The orange fluorescence disappeared when the A549 cells were pretreated with the lysosome inhibitor bafilomycin A1 prior to bleomycin exposure (photographs not shown), confirming its selective lysosomal localisation. The green fluorescence is emitted by the binding of acridine orange to nucleic acids. Anti-bromodeoxyuridine (BrdU) immunostaining of A549 cells treated with (h) or without (g)bleomycin, rinsed with phosphate-buffered saline, and then stimulated with 10% foetal calf serum (FCS) for 48 h. During the last 60 min of the stimulation period, the cells were pulse-labelled with BrdU. Treatment with bleomycin abolished the incorporation ofBrdU by cells stimulated with FCS (h), indicating that bleomycin causes irreversible growth arrest. These photomicrograph are representative of the results obtained from three experiments.

Quantitative analyses of cellular senescence in A549 cells treated with bleomycin. a) Staining for senescence-associated β‐galactosidase activity. A549 cells were treated with or without various concentrations of bleomycin for 96 h or b) with (•) or without (○) 50 µg·mL−1 of bleomycin for various periods of time. c) Size of A549 cells treated with or without 50 µg·mL−1 of bleomycin for 120 h. d) Incorporation of bromodeoxyuridine (BrdU) by A549 cells. A549 cells were treated with or without various concentrations of bleomycin for 120 h, rinsed with phosphate-buffered saline and incubated with 0.1% foetal calf serum (FCS; ○) or 10% FCS (•) for 48 h. During the last 60 min of the incubation, the cells were pulse-labelled with 10 µM of BrdU; cells that had incorporated BrdU were detected using immunocytochemistry. **: p<0.01 versus cells not treated with bleomycin. All data are presented as mean±sem for four samples. The results are representative of of two out of three experiments.
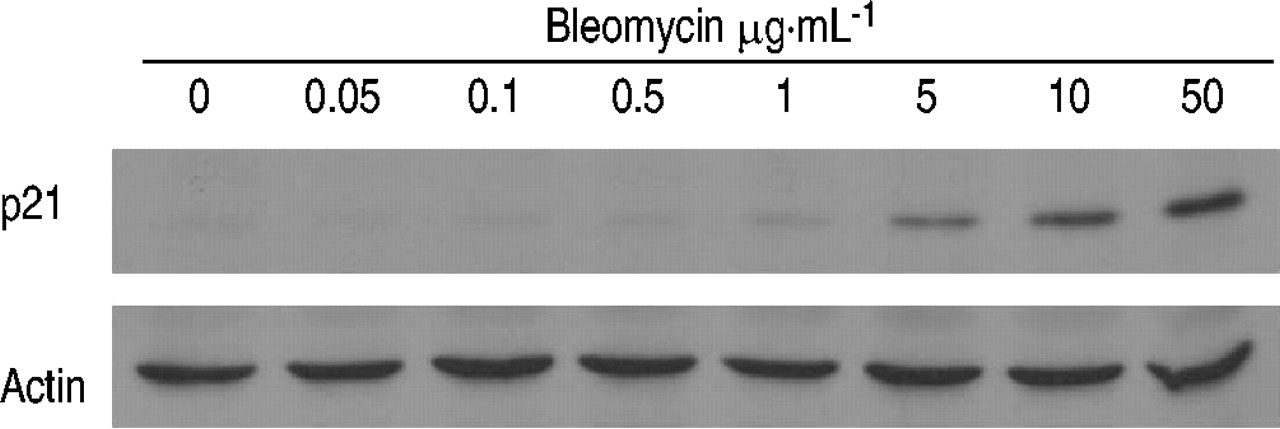
An immunoblot analysis of p21CIP1/WAF1/Sdi1 protein after treatment with bleomycin. A549 cells were treated with various concentrations of bleomycin for 120 h. The cell lysate (50 µg per lane) was analysed using immunoblotting with anti-p21 or anti-actin antibodies. The results are representative of two experiments.

a) Senescence-associated β‐galactosidase activity and b, c) cell morphology in primary rat type II cells treated with or without bleomycin (5 µg·mL−1) for 24 h (b shows untreated cells and c bleomycin-treated cells); **: p<0.01 versus cells not treated with bleomycin. Data are presented as mean±sem for four samples. The results are representative of two experiments.
Intratracheal injection of bleomycin causes an increase in senescence-associated β‐galactosidase activity in alveolar epithelial cells in vivo
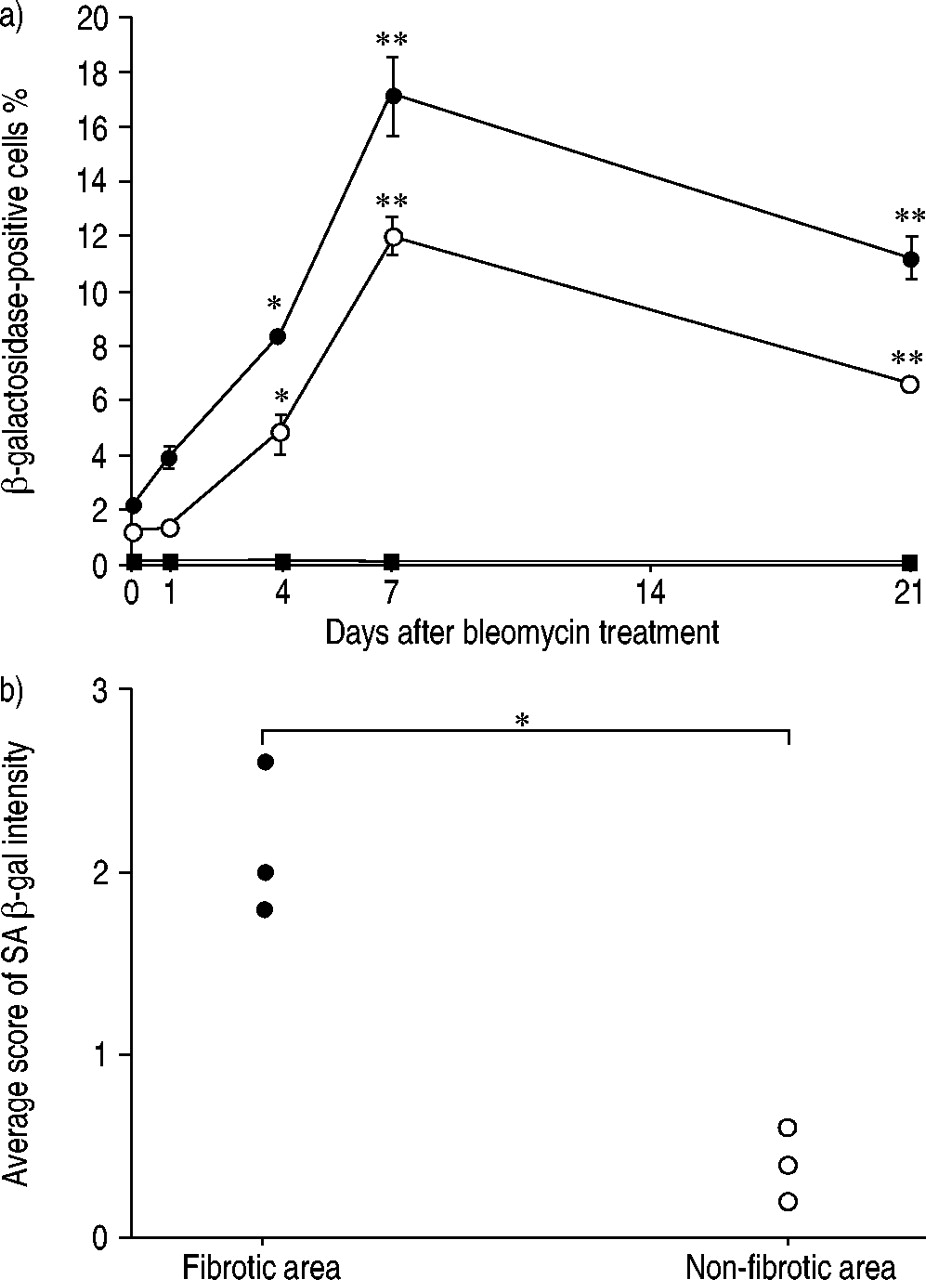
To determine whether bleomycin induces cellular senescence in alveolar epithelial cells in vivo, lung tissue sections obtained from mice that had received an intratracheal injection of bleomycin were examined using SA β‐gal staining. Before the bleomycin injection (day 0), SA β‐gal staining was almost negative in lung cells (fig. 5a and 5f⇓) other than alveolar macrophages (fig. 5n⇓), which are enriched with lysosomal β‐galactosidase 20. After the bleomycin injection, the number of lung cells that stained positive for SA β‐gal increased at day 4 or thereafter (fig. 5c–5e, 5h–5j⇓). The SA β‐gal-stained cells tended to be localised within collapsed or fibrotic lung lesions (fig. 5i and 5j⇓). An immunohistochemical analysis using anti-pancytokeratin, anti-SP‐A, anti‐CD31, and anti‐CD45 demonstrated that the SA β‐gal-stained cells were alveolar epithelial cells (fig. 5k and 5l⇓), particularly type II cells (fig. 5m⇓), and alveolar macrophages (fig. 5o⇓), but not endothelial cells (photographs not shown). As expected, the SA β‐gal-stained cells had not incorporated BrdU at any of the time points examined (fig. 5p⇓). After injection of PBS alone, the number of cells positive for SA β‐gal did not increase (data not shown). A semi-quantitative analysis also showed that the incidence of alveolar epithelial cells and type II cells positive for SA β‐gal staining reached a maximum at day 7 and an increase in the incidence of the positive cells was maintained for at least 21 days (fig. 6a⇓). In this analysis, SP-A positive cells may have in addition included nonciliated bronchiolar cells which also express SP‐A 21. In lungs ofmice at day 21 after bleomycin treatment, the score of SA β‐gal intensity was much higher in the fibrotic area than in the nonfibrotic area (fig. 6b⇓), suggesting that cellular senescence may be involved in pulmonary fibrogenesis.

Senescence-associated β‐galactosidase (SA β‐gal) activity in lung tissue sections from mice sacrificed before (a, f, n) and at 1 day (b, g), 4 days (c, h), 7 days (d, i, k, l, m, o, p), and 21 days (e, j) after an intratracheal injection of bleomycin (5 U·kg−1 body weight). At 4 h prior to sacrifice the mice received an intraperitoneal injection of 30 µg of bromodeoxyuridine (BrdU). a)–e) show SA β‐gal staining (blue colour). f)–j) show SA β‐gal staining followed by antilaminin immunostaining (brown colour) to identify the alveolar structure. k) and l) show SA β‐gal staining followed by antipancytokeratin immunostaining (brown colour) to identify the epithelial cells. Cell nuclei were stained with a nuclear fast red solution. m) shows SA β‐gal staining followed by anti‐SP‐A immunostaining (brown colour) to identify the type II cells, the cells nuclei were stained with a nuclear fast red solution. n) and o) show SA β‐gal staining followed by anti‐CD45 immunostaining to identify haematopoietic cells (brown colour). Cell nuclei were stained with a nuclear fast red solution. Intraluminal alveolar macrophages are positively stained for SA β‐gal. p) SA β‐gal staining (black arrows) followed by anti-BrdU immunostaining (white arrows) shows that the SA β‐gal-stained cells do not incorporate BrdU.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Semi-quantitative analyses of senescence-associated β‐galactosidase (SA β‐gal) activity in lung tissue sections. a) Time course of SA β‐gal activity in lung tissue sections from mice sacrificed before (day 0) and at days 1, 4, 7, and 21 after an intratracheal injection of bleomycin (•: cells positive for both SA β‐gal activity andanti‐SP‐A immunostaining; ○: cells positive for both SA β‐gal activity and antipancytokeratin immunostaining; ▪: cells positive forboth SA β‐gal activity and anti‐CD31 immunostaining). All data are mean±sem for three mice at each time point. *: p<0.05, **: p<0.01 versus day 0. b) The average grade of SA β‐gal intensity in relation to fibrosis in lungs of mice at 21 days after an intratracheal injection of bleomycin. Individual data from three mice are indicated. *: p<0.05.
Discussion
Although recent evidence indicates that epithelial regeneration is impaired in pulmonary fibrosis, the responsible mechanism is unknown 4. The present study demonstrated that treatment with bleomycin induces a phenotype that is indistinguishable from that of senescence in alveolar epithelial cells in vitro and in vivo. These results suggest that cellular senescence may be responsible for the impaired epithelial regeneration that occurs following bleomycin-induced lung injury.
Cellular senescence is a state of irreversible growth arrest which is characterised by distinct morphological and biochemical changes 9, 10. This state is induced either by telomere shortening or by telomere-independent signals, such as DNA damage and oxidative stress 9. The present study using alveolar epithelial cells supports previous findings showing that DNA damaging agents, such as bleomycin, actinomycin D, and etoposide, induce a senescence phenotype in normal fibroblasts and some tumour cell lines 21.
Regeneration of the injured epithelium involves the proliferation of alveolar type II cells. After an intratracheal injection of bleomycin, the early death of alveolar type I cells is followed by the attempted proliferation of type II cells 22. However, the proliferative capacity of the type II cells following bleomycin-induced injury is seriously affected, resulting inthe absence of appropriate re-epithelialisation; the lack of re-epithelialisation allows activated fibroblasts to migrate, through defects in the epithelium, from the interstitial in the intraluminal compartments 22. This pathway of impaired epithelial cell regeneration leading to pulmonary fibrosis is supported by previous findings showing that the enhancement of epithelial proliferation by treatment with keratinocyte growth factor or hepatocyte growth factor ameliorates bleomycin-induced pulmonary fibrosis in mice 23, 24. These findings suggest that the proliferation of alveolar epithelial cells is important for preventing the progress of bleomycin-induced pulmonary fibrosis. In this context, the present study suggests that the proliferation of alveolar epithelial cells may be limited as a result of cellular senescence, providing a new insight into the mechanism of pulmonary fibrosis.
The induction of cellular senescence does not indicate thatcellular functions are depressed. For example, many studies have shown that senescent cells produce higher levels of matrix metalloproteinases, such as collagenases and stromelysin, and profibrotic cytokines, such as interleukin‐1β andtransforming growth factor‐β (TGF‐β), than normal cells 25–27. These studies suggest that senescent cells may be involved not only in impaired tissue regeneration but also in the inflammatory response and extracellular matrix remodelling. Future studies are needed to examine whether senescence of alveolar epithelial cells is associated with functional abnormalities.
The limitation of this study is that the authors used A549 cells, immortalised cancer cells that lack the proliferative controls found in nontransformed cells. This notion is important because bleomycin is an anticancer drug which causes DNA damage in cancer cells. It is therefore difficult to extrapolate the present studies in vitro findings with A549 cells to normal alveolar epithelial cells. However, the authors found that treatment of primary rat type II cells with bleomycin also induced a senescence phenotype characterised by an increased SA β‐gal activity and a flat and enlarged cell morphology. The authors also found that an in vivo exposure of bleomycin in mice caused an increase in SA β‐gal activity in normal alveolar epithelial cells, suggesting that the response of A549 cells to bleomycin is not very different from that of normal alveolar epithelial cells.
The second limitation of this study is that the authors used an SA β‐gal staining method to identify senescent cells in lung tissue 17. This method has been widely used by many investigators who have shown a good correlation between SAβ‐gal activity and the onset of senescence in vitro and invivo 12, 13, 15. However, SA β‐gal activity may not be specific for senescence in all tissues, as it may also represent the expression of normal endogenous lysosomal acid β‐galactosidase and may not distinguish senescent cells from quiescent or terminally differentiated cells 16, 28. In fact, the current authors found that alveolar macrophages in normal murine lungs also exhibits SA β‐gal activity (fig. 5n⇑), suggesting that this marker must be used cautiously. The current authors also do not exclude the possibility that bleomycin may directly affect the SA β‐gal activity. The expression of SA β‐gal may also be different in rodent and human cells. An alternative method of detecting senescent cells in vivo would be the determination of telomere length by in situ hybridisation. However, this method is not suitable for the detection of telomere-independent senescence induced by DNA damage and oxidative stress, in the case of bleomycin. Thus, an absolute marker enabling the specific identification of cellular senescence in vivo, particularly stress-induced senescence, is not yet available. In this context, the present authors do not completely exclude the possibility that SA β‐gal activity might also reflect cell conditions other than senescence.
The third limitation of this study is that although bleomycin-induced pulmonary fibrosis in rodents has often been used as a model of human pulmonary fibrosis, this model does not resemble the histological and physiological features of human idiopathic pulmonary fibrosis (IPF). For example, a recent study showed that the early stages of bleomycin-induced lung injury in rats mimic diffuse alveolar damage (DAD), whereas the later, more chronic stages exhibit focal peribronchialar inflammation and fibrosis associated with paracicatrical emphysematous changes 29. In humans, bleomycin-induced lung injury has been shown toresemble DAD or bronchiolitis obliterans organising pneumonia 29, but not IPF. Thus, the bleomycin-induced model resembles only a particular group of interstitial lungdiseasesin humans, mainly those associated with peribronchovascular involvement 30. Therefore, extrapolation of the present findings in bleomycin-induced pulmonary fibrosis tothe situation in humans needs to be done with great caution.
Although the present study has several limitations, the current authors propose that epithelial senescence may be involved in bleomycin-induced fibrosis and possibly, in other types of pulmonary fibrosis. In pulmonary fibrosis, repeated deoxyribonucleic acid (DNA) and oxidative damage promotes epithelial cell senescence, thereby limiting their capacity to proliferate. The continuous proliferation of epithelial cells required to regenerate the damaged epithelium also accelerates telomere shortening and promotes epithelial cell senescence. When the epithelial cells reach the senescence stage, epithelial regeneration ceases and fibrosis progresses. This model provides a plausible explanation for the chronic nature of pulmonary fibrosis, which evolves slowly over many years. A detailed characterisation of cellular senescence in lung tissue samples should help to provide a better understand the pathogenesis of pulmonary fibrosis, and possibly, to identify new targets for treatment.
Acknowledgments
The authors are very grateful to M. Shino and Y. Sugimura for their excellent technical assistance.
- Received February 4, 2003.
- Accepted April 30, 2003.
- © ERS Journals Ltd
References









